
Capítulo de livro publicado no livro do I CONGEB. Para acessa-lo clique aqui.
DOI: https://doi.org/10.53934/9786585062046-17
Este trabalho foi escrito por:
Maria Júlia de França Souza Silva* ; Alexandre Freitas da Silva ; Gabriel da Luz Wallau
*Maria Júlia de França Souza Silva – [email protected]:
Com o passar dos anos, as técnicas de bioinformática vem deixando de ter um papel de complementaridade em diversos estudos, para ter um de fundamentalidade no mesmo. As infinitas possibilidades que essa área da biologia molecular traz para a comunidade científica é imensurável e vem sendo muito bem aproveitada. Os Culicídeos, mais popularmente conhecidos como “mosquitos” ou “pernilongos”, são de grande importância médica pela sua capacidade de transmitir patógenos para humanos e animais, tendo uma distribuição em escala global. Por conta disto, eles caracterizam um grupo de organismos de grande interesse mundial e se faz necessário a realização de investigações sobre sua biologia e sobre sua história evolutiva, para que assim, sejam criadas estratégias de controle mais adequadas na tentativa de diminuir as sequelas deixadas por eles. Portanto, esta revisão visa mostrar a importância da família Culicidae, associando com a progressão da bioinformática e suas principais estratégias aplicadas aos Culicídeos, e por último alguns dos resultados que foram obtidos através dessas técnicas ao longo dos anos.
Palavras–chave: bioinformática; biologia molecular; filogenia; genomas; mitogenomas
INTRODUÇÃO
A família Culicidae faz parte da ordem Diptera e compreende mais de 3 mil espécies de mosquitos encontrados em todas as regiões do mundo, mas ocorrendo principalmente em áreas tropicais e temperadas. Os Culicídeos são divididos em duas subfamílias: Culicinae e Anophelinae, que possuem 110 e 3 gêneros, respectivamente, apesar de existirem inconsistências entre os autores em relação às classificações dos táxons. Na subfamília Culicinae encontramos 11 tribos e alguns dos seus gêneros são: Aedes, Sabethes, Culex, Mansonia e Coquillettidia. Já a Anophelinae, caracteriza uma quantidade menor de táxons onde estão alocados somente 3 gêneros: Anopheles, Bironella e Chagasia.
Várias espécies são de interesse médico e veterinário por serem vetoras de diversos patógenos, como vermes filariais, protozoaŕios do gênero Plasmodium e diversos vírus, como por exemplo o vírus Zika. Em um estudo realizado em 2018 estimou-se que em torno de 725 mil pessoas perderam suas vidas devido a ação de mosquitos vetores ao redor do mundo, como o Aedes aegypti e Anophele gambie (1). Por conta disso, os Culicídeos são fortemente estudados no mundo todo através de diversas abordagens, dentre elas está o estudo da evolução e da filogenia desta grande família. A melhor compreensão das relações filogenéticas dentro desse grupo é fundamental para compreender possíveis novas ameaças e possíveis novos vetores a partir da análise de semelhanças evolutivas entre as espécies. A sistemática tem uma origem muito antiga, desde Aristóteles já temos registros de tentativas de organização e separação daquilo que era visto. Contudo é a partir de Linné e seu sistema binominal que uma melhor organização desses estudos se tornou possível. Inicialmente, as análises comparativas eram totalmente baseadas em métodos observativos e morfológicos a fim de separar e agrupar as espécies. Com o advento da tecnologia e todas as descobertas da biologia celular e molecular foi possível começar a utilizar dados biológicos para inferir esses relacionamentos de forma mais precisa e confiável.
A bioinformática é uma área recente na biologia, mas que já promoveu diversas descobertas relevantes para o campo e dentre estas, mudanças filogenéticas de diversos grupos de organismos trazendo novas perspectivas. Hoje, temos acesso a uma vasta rede de programas e softwares que nos dão a chance de processar, organizar, analisar e estudar esses dados biológicos, desde o DNA até às proteínas.
O objetivo desta revisão é trazer a versatilidade da bioinformática e os principais achados que ela proporcionou para o estudo da evolução e biologia do grupo dos Culicídeos, que são de grande importância através de uma revisão pelas técnicas utilizadas e pelas diferentes abordagens que podem ser adotadas. Com isso, também trazer a importância de dados básicos sobre vetores e suas relações interespecíficas para o melhor entendimento de possíveis dinâmicas ecológicas e vetoriais, e assim, colaborar para para o desenvolvimento de estratégias de controle.
TÓPICOS
Primeiros estudos:
No ano de 1951, se deu o início de estudos evolutivos da família Culicidae a partir da construção da primeira filogenia evolutiva realizada por Ross (2). As primeiras sistematizações eram bem básicas se comparados aos método disponíveis atualmente, sendo possível apenas separar as três subfamílias: Anophelinae, Culicinae e Toxorhynchitinae, propostas nesta época. Existem importantes autores na cladística dos Culicídeos, dentre eles Forattini, Lane e Judd que contribuíram com a construção de suas chaves de identificação utilizadas até hoje para diversos gêneros dentro da família. Esses estudos examinavam as diferentes fases do ciclo de vida dos mosquitos, larva (que possui quatro estágios), pupa e mosquito adulto. Os estudos evoluíram de utilizar somente a observação e desenhos ilustrativos para a organização sistemática para uso de cladogramas de parcimônia a partir da década de 70 (3).
Os estudos morfológicos e separação de táxons a partir da observação dessas características se baseia em características subjetivas que estão sujeitas a diferentes interpretações levando a uma série de inconsistências no momento da classificação. Um problema bem pontual dentro desse grupo de organismos relacionado a sua classificação morfológica é a existência de alguns complexos de espécies, como por exemplo o complexo Culex pipiens que inclui várias subespécies, como Cx. quinquefasciatus, Cx. molestus e Cx. pallens. Esse complexo é de extrema importância médica e veterinária devido a capacidade dessas espécies agirem como vetores de diversos patógenos, como: vírus do Nilo Ocidental (WNV), vírus da febre do Vale Rift (RVFV) e vírus Síndbis (SINV) (4). Logo, é extremamente importante que as espécies sejam devidamente identificadas e separadas para que o estudo e desenvolvimento de tecnologias de controle vetorial sejam construídas com a melhor acurácia possível.
Para isso, com o passar do tempo os estudos morfológicos foram sendo combinados com novas metodologias baseadas em caracteres genéticos com o objetivo de obter resultados mais precisos (5). Contudo, além de ser possível utilizar o DNA do núcleo celular para investigação evolutiva, também é possível utilizar o DNA mitocondrial, que é um conjunto genético diferente do nuclear e que pode trazer outras informações. Isto se dá pelas características encontradas no DNA mitocondrial, como seu alto número de cópias e um número bem maior de mutações. Essa maior taxa mutacional do DNA mitocondrial o torna um marcador mais informativo para discernir diferentes espécies (6). O gene Citocromo Oxidase 1 (COX 1), por um exemplo, é um dos marcadores mitocondriais mais amplamente usado para identificação e sistemática molecular em diversos grupos como Nematoda e Arthropoda (7)(8).
Bioinformática e sua multifuncionalidade:
Muitos estudos consideram que o acontecimento que marcou o início da história da bioinformática ocorreu em 1953 a partir do modelo de dupla hélice do DNA proposto pelos pesquisadores James Watson e Francis Crick. Isso porque foi a partir deste entendimento mais completo da estrutura molecular da molécula responsável por carregar nossas informações genéticas foi possível ampliar diferentes abordagens de estudos e aplicar esse conhecimento para outras biomoléculas. Com o avanço das tecnologias, a área da biologia não ficou de fora quando se trata de inovações facilitadoras de estudo e que abrem um leque de possibilidades. A partir da década de 70, temos o surgimento de uma ferramenta que revolucionaria a área da biologia molecular, a criação dos métodos de sequenciamento de DNA. As primeiras formas foram elaboradas por Allan Maxam e Walter Gilbert (sequenciamento químico) e por de Alan Coulson e Frederick Sanger (sequenciamento de Sanger). Dessa forma, junto com essa oportunidade de organizar o material genético e estudá-lo melhor veio também a enorme quantidade de informação sendo produzida das mais variadas espécies gerando uma necessidade de organização e mineração deste volume de dados.
Com isso, a bioinformática surge sendo um campo multidisciplinar, empregando matemática e computação para extrair informações de dados biológicos em larga escala. Uma ferramenta bastante vantajosa foi a criação dos bancos de dados online, onde é possível ter acesso a inúmeras informações biológicas disponíveis para qualquer tipo de organismos. Podemos encontrar bancos de dados exclusivos para alguma informação, como por exemplo, o UniProt que caracteriza um banco específico para sequências proteicas, como também bancos de dados mais amplos contendo sequências proteicas e nucleotídicas como o GenBank. O desenvolvimento de algoritmos e de programas com a capacidade de trabalhar com dados biológicos também é uma parte fundamental dentro dessa aŕea.
Inúmeros programas foram criados nesses últimos 10 anos auxiliando na construção de análises filogenéticas e assim permitindo um melhor entendimento das relações intra e interespecíficas. Desses, os mais utilizados como o Geneious Prime, MITObim e BEAST permitem uma fácil análise e interpretação dos dados, além de permitir uma variedade de abordagens para um único set de informações. O Geneious Prime funciona como um programa multifuncional onde é possível visualizar e estudar sequências anotadas de DNA e RNA, realizar alinhamentos, simulação de clonagem molecular e análises filogenéticas. O programa BEAST (Bayesian Evolutionary Analysis Sampling Trees) é uma ferramenta extensamente utilizada para a reconstrução filogenética e a criação de hipóteses evolutivas (9). Seu algoritmo se destaca por construir as árvores baseadas na Cadeia de Markov Monte Carlo, que é baseada em uma distribuição probabilística, e por ter diversos modelos de relógio moleculares, que podem se adequar ao tipo específico de conjunto de dados levando em consideração suas nuances e assim obter o melhor resultado possível (10). Além de também possuir a possibilidade de incluir informações temporais baseadas em registros fósseis, trazendo uma maior complexidade para as análises. Após o sequenciamento genômico, é necessário realizar a etapa de montagem desse genoma. Dentro da gama de programas desenvolvidos para essa função está o MITObim que é específico para reconstrução de genomas mitocondriais (11). Este programa objetiva reconstruir mitogenomas sem a necessidade de um genoma de referência da espécie estudada, usando no lugar disto informação genômica de uma espécie distante como ponto de partida. Quando se obtêm um novo mitogenoma o próximo passo é a anotação gênica, geralmente realizada no webserver MITOS. No geral, é esperado que os organismos possuam todos os 37 genes conservados em eucariotos: 13 genes codificadores de proteínas, 22 RNAs transportadores e 2 RNAs ribossomais em uma ordem e estrutura padronizada. Contudo, através de um estudo realizado com espécies da tribo Sabethini foi registrado a primeira translocação gênica onde os pesquisadores notaram que dois tRNAs, Y e C, não estavam localizados entre o gene Citocromo Oxidase e o tRNA-W, mas sim entre tRNA-I e a região de AT de controle (30). Essa translocação foi colocada como sendo um autapomorfia das espécies de Sabetines encontrados na região Neotropical, ou seja, uma característica que foi derivada e vai ser encontrada exclusivamente dentro de um táxon terminal dentro da filogenia, podendo ser usado com critério de distinção entre os táxons dessa tribo.
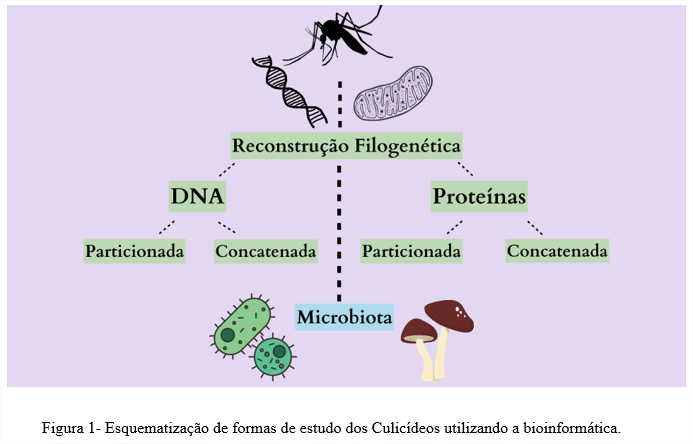
A partir da utilização desses programas é possível colocar em prática diferentes abordagens para a análise genômica evolutiva. Inicialmente, existem dois tipos de dados que podem ser utilizados: ácidos nucleicos (DNA e RNA) e os aminoácidos (proteínas). Cada grupo vai nos fornecer informações e visões diferentes do processo evolutivo que ocorreu ao longo do tempo dentro de um determinado grupo. As análises nucleotídicas a partir de genomas mitocondriais são mais estudadas e realizadas, e se mostraram bastante eficientes em reconstruções intraespecíficas dentro de alguns grupos, como os Diptera (12). Por outro, as análises baseadas em códons desse genoma podem trazer questões relevantes e singulares. Como os genes codificadores de proteínas possuem uma alta taxa de mudança em seus códons, principalmente na terceira posição, e essa substituição na grande maioria das vezes não caracteriza um aminoácido diferente, isso pode ser usado como medidor de taxa evolutiva (13). Além do mais, a utilização de sequências de proteínas são ideais em estudos que possuem o objetivo de caracterizar ramos mais antigos dentro do grupo de organismos estudados, sendo um conjunto de dados mais confiável que os nucleotídeos neste contexto. Também é possível realizar análises particionadas, onde cada dado será analisado separadamente, e que pode ser usada para as proteínas e os nucleotídeos de formas diferentes para melhor explorar o dado filogenético levando em consideração a diferença na taxa evolutiva de cada região do genoma.
Por último, é importante também ressaltar que a bioinformática nos permite ir mais fundo do que somente as análises filogenéticas, podendo combinar estas com estudos de viroma e microbioma, visando a caracterização global de vírus e outros microorganismos respectivamente. Assim como qualquer organismo, os mosquitos dispõe de uma série de microorganismos em seus sistemas e que vão ter um papel chave no seu ciclo de vida e nas suas respostas a estímulos e interações externas com o ambiente. Tanto bactérias quanto fungos podem ser encontrados, porém, as bactérias formam o grupo de maior quantidade e participação nesses processos. Esses estudos são extremamente relevantes, visto que as interações desses microrganismos e dos mosquitos ao longo do tempo podem também ter tido influência em seus comportamentos fisiológicos referentes à transmissão de arbovírus(14-18). O microbioma pode ser encontrado não somente no trato digestivo, mas também vai estar presente nas glândulas salivares, local chave para a competência vetorial do mosquito e sua possível transmissibilidade. A composição desse bioma impacta no sistema imunológico dos mosquitos (positivamente ou negativamente), na sua alimentação e pode modular a infecção e propagação de patógenos. Uma forma bastante utilizada para realizar a identificação das espécies é através de barcoding, utilizando o gene 16S rRNA para bactérias e o gene 18S rRNA para fungos em técnicas de PCR e sequenciamento (19). A partir do conhecimento dessa microbiota é possível elaborar possíveis estratégias de controle biológico mais eficientes utilizando os próprios microrganismos já associados aos mosquitos.
Principais achados a partir do uso da Bioinformática na família Culicidae:
Como mencionado anteriormente, vários estudos de grupos de organismos se beneficiaram das técnicas de bioinformática e puderam obter resultados interessantes e esclarecedores sobre suas relações de parentesco e evolução. Dentre esses, a família Culicidae é bastante explorada e estudada visto sua importância médica e veterinária já citada acima. Um gênero bastante relevante dentro desta família é o gênero Anopheles, que possui várias espécies vetoras como An. funestus, An. stephensi, An. messeae e An. dacie, transmissoras da Malária em vários continentes como a Europa, África e América do Sul (20-22). Esse gênero é formado por complexos de espécies, onde somente através de uma análise genômica é possível diferenciar os táxons. Contudo, algumas espécies podem estar proximamente relacionadas a esses complexos, como achado para a espécie An. christyi que nas filogenias gerada por um estudo em 2021 demonstrou uma relação com o complexo de Anopheles gambie(s.l), sendo este último o complexo com as principais espécies transmissoras de Malária (23). Esse tipo de achado é extremamente relevante para planos de controle de patógenos e de vigilância para possíveis novos vetores em áreas endêmicas. Outro resultado muito interessante retirado a partir desse tipo de análise é a possível data de surgimento desta família e seus grupos dentro do contexto geológico e evolutivo. A partir de análises filogenéticas datadas realizadas por um estudo foi visto que a aparente origem da família Culicidae ocorreu em torno de 182 milhões de anos atrás a partir da observação do último ancestral comum desse grupo. Esse surgimento coincide com o período Jurássico que é marcado pelo início do afastamento da Pangeia, o que colaborou com o aumento de diversificação e especiação de animais e plantas (24).
A tribo Aedini compreende gêneros importantes por conta da sua capacidade vetorial como Aedes, vetor de Dengue (DENV) e Chikungunya (CHIKV), e Haemagogus, vetor da Febre Amarela (25). O estudo de filogenias datadas ajudam a compreender como esses táxons se diversificaram ao longo do tempo e possivelmente entender a origem do seu padrão vetorial e assim determinar outras possíveis origens. Além disso, muitos estudos vêm se dedicando a caracterizar o microbioma encontrado em Aedes em todos seus estágios de vida (ovo, larva, pupa e adulto) e assim verificar o possível impacto dele nas transmissões de patógenos e nos comportamentos biológicos desse táxon (26, 27). Através dos estudos metagenômicos proporcionados pela bioinformática foi possível caracterizar diversos gêneros de bactérias encontrados com um certo padrão dentro de Aedes, dentre esses estão: Asaia, Serratia, Klebsiella e Pseudomonas (27). O gênero de bactéria Asaia pode ser encontrado em diversas partes do corpo do mosquito e já foi identificado espécies de Anopheles vetoras de Plasmodium, como An. gambie e An. maculipennis mostrando que pode ser uma opção para o combate desse parasitoide. Além de que já foi observado microrganismos que agem estimulando o sistema imune do mosquito e assim bloquear ou aumentar a reação contra algum patógeno, como por exemplo, o fungo Beauveria bassiana aumenta a resistência Dengue (DENV) através de um aumento na atividade de vias imunológica muito importante desse grupo, a via Toll e a via JAK-STAT (28). Ademais, apesar da família Culicidae ser extensamente estudada há muitos anos, ainda existem diversas espécies pouco conhecidas e que nem sequer tiveram seu genoma caracterizado, logo, através do sequenciamento e montagem de genomas é possível realizar a descrição, e assim, incluir essas novas espécies em estudos e análises, e investigar melhor seus traços evolutivos e ecológicos. Um estudo realizou a primeira descrição do genoma mitocondrial das espécies Aedes vexans e Ochlerotatus caspius e através de reconstruções filogenéticas buscaram estabelecer possíveis marcadores para facilitar futuros estudos epidemiológicos que que incluam esses táxons (29).
CONCLUSÕES
Os avanços tecnológicos dos últimos 30 anos na área de sequenciamento e bioinformática expandiram substancialmente as funcionalidades e amplitude de possibilidades dentro das áreas da saúde e da biologia. Com o surgimento das técnicas de biologia molecular veio juntamente a possibilidade de explorar mais profundamente as moléculas responsáveis pelo desenvolvimento e pelos traços biológicos dos organismos, como o DNA e as proteínas. Apesar de ser um grupo que passa muitas vezes despercebido, os Culicídeos constituem uma família de insetos com um alto nível de interação com a população humana e que é responsável pela transmissão de patógenos causadores de diversos tipos de doenças por todo o globo.
O uso da taxonomia clássica é altamente utilizado até os dias de hoje, estudos morfológicos são feitos para a caracterização e diferenciação de diversas espécies e compõem uma parte fundamental e significativa da cladística. Contudo, é inegável que como qualquer tipo de estudo ela possui suas limitações e imprecisões que podem afetar diretamente a acurácia de certos estudos com grupos importantes de organismos, tais quais os Culicídeos. Por isso foi tão fundamental a criação e o agregamento de tecnologias ao longo dos últimos anos para unir a taxonomia clássica com a taxonomia baseada em dados moleculares e assim aumentar sua precisão e sua quantidade de informação dali retiradas, e consequentemente aplicadas a favor da ciência. A adição de dados moleculares em estudos filogenéticos permitiram que uma maior clareza fosse trazida para a classificação da família Culicidae, juntamente com dados geológicos, hipóteses e possíveis novos vetores.
Ademais, a aplicação de ferramentas bioinformáticas para a caracterização e estudo da composição do microbioma das espécies de Culicídeos é uma linha de experimentação tão importante quanto da filogenética. A partir dos estudos de microbiota foi possível caracterizar a existência de um microbiome “core” que caracteriza um padrão de grupos de bactérias e fungos encontrados em gêneros de Culicídeos de localidades diferentes. Isso somado com a crescente busca por organismos que podem ser utilizados como forma de controle – biológico visando uma diminuição de uso de químicos nocivos para a natureza, faz com que relevância e a potencialidade dessa microbiota seja colocada em destaque para utilizá-la ao nosso favor na luta contra as arboviroses. Essa aplicabilidade ainda pode ser muito explorada principalmente por praticamente termos transmissão anual em quase todos os estados brasileiros de Dengue, Zika e Chikungunya. Assim, seria interessante investigar se isso tem alguma ligação com o seu microbioma, ou, se o mesmo pode ser utilizado para combater número tão altos de contaminação por esses patógenos.
Assim, é notável como a bioinformatica nos permite ter uma liberdade e versatilidade em estudos e experimentos (Fig. 1) dentro desse grupo de mosquitos e vem trazendo resultados promissores e determinantes no melhor entendimento da sua evolução, distribuição e relação intraespecífica deles.
AGRADECIMENTOS
Gostaria de agradecer ao Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) pelo apoio através do Programa de Iniciação Científica. Ao Dr. Prof. Gabriel Wallau e ao Msc. Alexandre Freitas pelas correções e suporte durante a escrita desta revisão.
REFERÊNCIAS
- GZH Saúde. Mosquitos mataram mais de 725 mil pessoas ao redor do mundo em 2018, aponta estudo. 2022 [acesso em 2022 Julho 20]. Disponível em: https://gauchazh.clicrbs.com.br/saude/noticia/2022/07/mosquitos-mataram-mais-de-725-mil-pessoas-ao-redor-do-mundo-em-2018-aponta-estudo-cl5ld767c007p014sisdw7e81.html
- Ross H.H. Conflict with Culex. Mosquito News. 1951. Volume 11: 128–132
- Harbach R. The Culicidae (Diptera): a review of taxonomy, classification and phylogeny. Zootaxa. 2007. Volume 1668: 591–638.
- Turell M J. Members of the Culex pipiens Complex as Vectors of Viruses. Journal of the American Mos. Control Association. 2012. Volume 28: 123-126.
- Motta M. A, Lourenço-de-Oliveira R, Sallum M. A. M. Phylogeny of genus Wyeomyia (Diptera: Culicidae) inferred from morphological and allozyme data. T. Canadian E. 2007. Volume 139: 591-627.
- Sharma N, Pasala M. S, Prakash A. Mitochondrial DNA: Epigenetics and environment.Enviro and mol mutagenesis. 2019. Volume 60: 668-682.
- Dittrich‐Schröder G. U. D. R. U. N, Wingfield M. J, Klein H, Slippers B. DNA extraction techniques for DNA barcoding of minute gall‐inhabiting wasps. Mol Eco Resources. 2012. Volume 12: 109-115.
- Ngui R, Mahdy M. A, Chua K. H, Traub R, Lim Y. A. Genetic characterization of the partial mitochondrial cytochrome oxidase c subunit I (cox 1) gene of the zoonotic parasitic nematode, Ancylostoma ceylanicum from humans, dogs and cats. Acta Tro. 2013. Volume 128: 154–157.
- Suchard M A, Lemey P, Baele G, Ayres D L, Drummond A J, Rambaut A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus evo. 2018. Volume 4: 1-16.
- Reyes A, Alves J. M. P, Durham A, Gruber A. Use of profile hidden Markov models in viral discovery: Current insights. Adva in Genom and Genet. 2017. Volume 7: 29-45.
- Hahn C, Bachmann L, Chevreux B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—a baiting and iterative mapping approach. Nu acids research. 2013. Volume 41: e129-e129.
- Talavera G, Vila R. What is the phylogenetic signal limit from mitogenomes? The reconciliation between mitochondrial and nuclear data in the Insecta class phylogeny. BMC Evol Biol. 2011. Volume 11: 315-330.
- Inagaki Y, G B Simpson A, Dacks J B, Roger A J. Phylogenetic Artifacts Can be Caused by Leucine, Serine, and Arginine Codon Usage Heterogeneity: Dinoflagellate Plastid Origins as a Case Study. Syst Bio. 2004. Volume 53: 582–593.
- Hegde S, Rasgon J L, Hughes G L. The microbiome modulates arbovirus transmission in mosquitoes. Curr opin in viro. 2015. Volume 15: 97-102.
- Johnson K. N. The impact of Wolbachia on virus infection in mosquitoes. Viruses. 2015. Volume 7: 5705-5717.
- Shi C, Zhao L, Atoni E, Zeng W, Hu X, Matthijnssens, J, et al. Stability of the Virome in Lab- and Field-Collected Aedes albopictus Mosquitoes across Different Developmental Stages and Possible Core Viruses in the Publicly Available Virome Data of Aedes Mosquitoes. mSystems. 2020. Volume 5: e00640-20.
- Ricci I, Damiani C, Capone A, DeFreece C, Rossi , Favia, G. Mosquito/microbiota interactions: from complex relationships to biotechnological perspectives. Curr opin in micro. 2012. Volume 15: 278-284.
- Minard G, Mavingui P, Moro C V. Diversity and function of bacterial microbiota in the mosquito holobiont. Para & vect. 2013. Volume: 6: 1-12.
- Jackson E E, Hawes I, Jungblut A D. 16S rRNA gene and 18S rRNA gene diversity in microbial mat communities in meltwater ponds on the McMurdo Ice Shelf, Antarctica. Polar Biol. 2021. Volume: 44: 823–836.
- Naumenko A N, Karagodin D A, Yurchenko A A, et al. Chromosome and Genome Divergence between the Cryptic Eurasian Malaria Vector-Species Anopheles messeae and Anopheles daciae. Genes (Basel). 2020. Volume 11 165-187.
- Rodriguez A M, Hambly M G, Jandu S, Simão-Gurge R, Lowder C, Lewis E E, et al. Histamine ingestion by Anopheles stephensi alters important vector transmission behaviors and infection success with diverse plasmodium species. Biomol. 2021. Volume: 11: 719-742.
- Yokoly F N, Zahouli J, Small G, Ouattara A F, Opoku M, de Souza D K, et al. Assessing Anopheles vector species diversity and transmission of malaria in four health districts along the borders of Côte d’Ivoire. Mala jour. 2021. Volume 20: 409-420.
- Ghassemi-Khademi T, Oshaghi M A, Vatandoost H, Madjdzadeh S M, Gorouhi, M A. (2021). Utility of Complete Mitochondrial Genomes in Phylogenetic Classification of the Species of Anopheles (Culicidae: Anophelinae). Jour of arthro-borne disea. 2021. Volume: 15: 1–20.
- da Silva A F, Machado L C, de Paula M B, da Silva Pessoa Vieira C J, de Morais Bronzoni R V, de Melo Santos M A V, et al. Culicidae evolutionary history focusing on the Culicinae subfamily based on mitochondrial phylogenomics. Scient repor. 2020. Volume 10: 1-14.
- Cano M E, Marti G, Balsalobre A, Muttis E, Bruno E, Rossi G C, et al. Database of Sabethes and Haemagogus (Diptera: Culicidae) in Argentina: Sylvatic Vectors of the Yellow Fever Virus. 2021. Jour of Med Entomo. Volume 58: 1762 – 1770.
- Scolari F, Sandionigi A, Carlassara M, Bruno A, Casiraghi M, Bonizzoni M. Exploring changes in the microbiota of Aedes albopictus: comparison among breeding site water, larvae, and adults. 2021. Front in Micr. Volume 12: 62417.
- Scolari F, Casiraghi M, Bonizzoni M. Aedes spp. and their microbiota: a review. Front in Micro. 2019. Volume 10: 2036.
- Caragata E P, Tikhe C V, Dimopoulos G. Curious entanglements: interactions between mosquitoes, their microbiota, and arboviruses. Curr opin in virol. 2019. Volume 37: 26-36.
- Ma X X, Wang F F, Wu T T, Li Y, Sun X J, Wang C R, et al. First description of the mitogenome and phylogeny: Aedes vexans and Ochlerotatus caspius of the Tribe Aedini (Diptera: Culicidae). Infecion, genet and evol: journal of molecular. 2022. Volume 102: 105311.
30. Lorenz C, Alves J M P, Foster P G. et al. First record of translocation in Culicidae (Diptera) mitogenomes: evidence from the tribe Sabethini. BMC Genom. 2019 Volume 20: 1-8.

{kind=link}