
Capítulo de livro publicado no livro do I CONGEB. Para acessa-lo clique aqui.
DOI: https://doi.org/10.53934/9786585062046-3
Este trabalho foi escrito por:
Ana Rafaela Carvalho Monte1*; Ana Carolina Lima Camargo2 ; Gabrielle Maria Giovanna da Silva Gonçalves; Thiago Adalton Rosa Rodrigues ; Júlia Nicoliello Pereira de Castro; Sueli Matilde da Silva Costa; Mônica Barbosa de Melo1
Resumo: A anemia falciforme (AF) é a doença mais comum dentre as doenças falciformes (DF) que são caracterizadas por uma mutação de ponto no gene que codifica a globina beta da hemoglobina. As complicações oftalmológicas podem ocorrer nas DF e incluem anormalidades na conjuntiva, infartos orbitários, retinopatia e hemorragia retiniana. A retinopatia falciforme (RF) ocorre devido à vaso-oclusão da microvasculatura ocular, podendo levar a um processo de neovascularização que eventualmente se desdobra em hemorragia vítrea e descolamento de retina causando perda de visão. A RF é dividida em não proliferativa (RNP) e proliferativa (RFP). A RFP pode afetar até 20% dos pacientes. Esta revisão tem como objetivo apresentar a relação da retinopatia falciforme com os genes Roundabout Guidance Receptor 1 (ROBO 1) e RAB38, Member RAS Oncogene Family, uma vez que estão associados a angiogênese e proliferação celular, vias importantes para o desenvolvimento da doença. Até o momento, há poucos estudos relacionando os genes RAB38 e ROBO1 com a retinopatia falciforme, principalmente na sua forma proliferativa. Sendo assim, esta revisão apresenta uma visão dos principais mecanismos moleculares desses genes associados à retinopatia falciforme proliferativa.
Palavras–chave: Anemia falciforme, expressão gênica,Retinopatia Falciforme.
- INTRODUÇÃO
As doenças falciformes (DF) são um grupo de hemoglobinopatias hereditárias caracterizadas por uma mutação no gene que codifica a subunidade β-globina. Nesse grupo de DF, a anemia falciforme (AF) é a mais comum, sendo uma doença autossômica recessiva hereditária, causada pela substituição de uma base nitrogenada adenina por timina (GAG – GTG) no códon 7 do cromossomo 11p15.5.7, da β -globina (1).
Essa substituição resulta na troca do aminoácido ácido glutâmico por valina, dando origem a uma hemoglobina alterada denominada hemoglobina S (HbS) (2). Quando ocorre a substituição nos dois alelos (homozigose), origina-se a anemia falciforme. Já no traço falciforme o indivíduo heterozigoto apresenta um único gene de hemoglobina afetado (HbS) sendo o outro gene normal (HbA), ocorrem manifestações leves ou até mesmo não observáveis (1).
Na anemia falciforme, durante a desoxigenação que ocorre com a passagem dos eritrócitos pela microcirculação, a substituição do ácido glutâmico hidrofílico pela valina hidrofóbica resulta na interação com outros resíduos também hidrofóbicos na cadeia da β-globina de outra molécula de HbS desoxigenada3. Essa interação origina polímeros que se alongam em fibras helicoidais. Os polímeros, quando agrupados, enrijecem e fazem com que as células desenvolvam o formato de foice, além de ativar cascatas de várias outras anormalidades celulares. Devido a polimerização, ocorrem eventos de vaso-oclusão bem como anemia hemolítica devido à fragilidade dos glóbulos vermelhos (1).
A polimerização da hemoglobina S é reversível quando no início, mas conforme as repetições dos ciclos de desoxigenação ocorrem, as alterações nas bicamadas lipídicas e nas proteínas da membrana eritrocitária começam a interferir em vários outros aspectos da fisiologia das hemácias (3). Esses numerosos eventos levam ao aumento da hemólise e a interação dos eritrócitos com as células endoteliais, leucocitárias e plaquetárias (1). A hemólise tem como resultado a liberação da hemoglobina livre na circulação levando a redução do óxido nítrico, substância vasodilatadora, produzida pelas células endoteliais que regula o tônus vascular (3). A diminuição da disponibilidade do óxido nítrico contribui para a vasoconstrição favorecendo potencialmente a vaso-oclusão e, portanto, participando da fisiopatogênese de manifestações clínicas da doença falciforme (1).
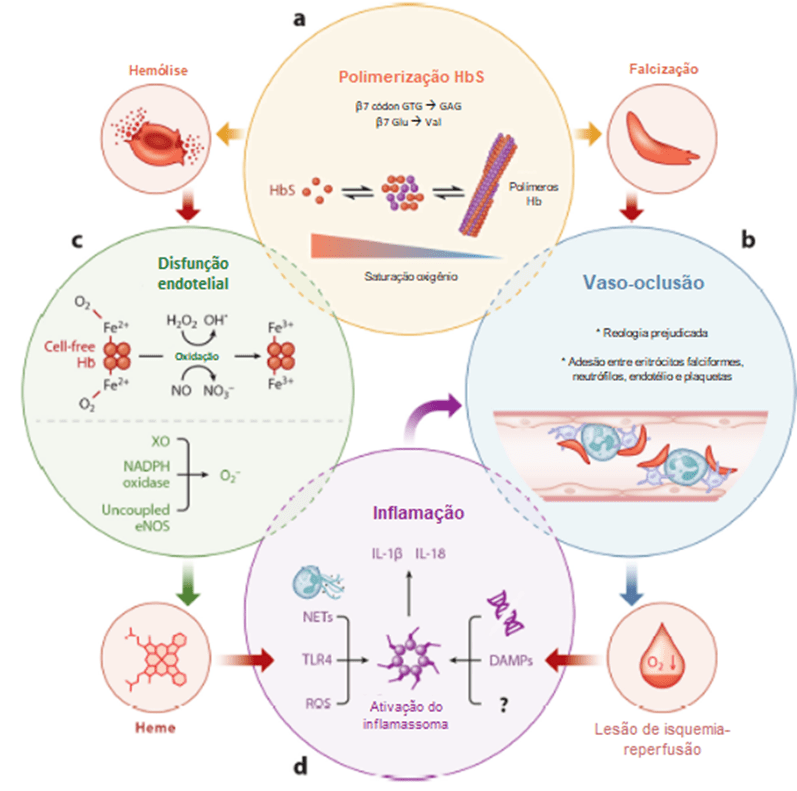
Figura 01. Fisiopatologia molecular da doença falciforme. (a) Polimerização da Hemoglobina. Um polimorfismo de um único nucleotídeo no gene da β-globina acarreta na substituição de ácido glutâmico por valina na sétima posição na cadeia da β-globina. Após a desoxigenação, as moléculas de hemoglobina alterada (HbS) polimerizam para formar feixes. Os feixes de polímeros resultam em falcização de eritrócitos. (b) reologia prejudicada do sangue e agregação de eritrócitos falciformes com neutrófilos, plaquetas e células endoteliais para promover a estase do fluxo sanguíneo, conhecido como vaso-oclusão e resulta em lesão de isquemia-reperfusão. Os feixes de hemoglobina (Hb) também promovem a hemólise ou lise dos eritrócitos, liberando Hb livre de células na circulação sanguínea, sendo uma Hb oxigenada (Fe2+3–) e metemoglobina (Fe3+). (c) Isso resulta em Hb livre entrando na corrente sanguínea. A hemoglobina pode reagir com o peróxido de hidrogênio através da reação de Fenton para formar o radical livre hidroxila – também chamado HO2 e a metemoglobina, que é o ferro no estado Fe3+. Além disso, NADPH oxidase, xantina oxidase (XO) e óxido nítrico desacoplado endotelial (eNOS) geram radicais livres de oxigênio para promover a disfunção endotelial. A metemoglobina (Fe3+) é degradada para liberar heme livre, um dos principais padrões moleculares associados ao dano dos glóbulos vermelhos (DAMP); (d) Produção de espécies reativas de oxigênio (ROS), ativação do receptor TLR4, produção de armadilha de neutrófilos extracelular (NET), liberação de DAMPs derivados de tecidos ou células, DNA e outros fatores desconhecidos. A lesão da heme livre ou I-R pode contribuir para a inflamação estéril, ativando a via do inflamassoma em vasos sanguíneos e células inflamatórias para liberar IL-1β. Finalmente, a inflamação estéril promove ainda mais a oclusão vascular por meio de uma alça de retroalimentação que promove a adesão de neutrófilos, plaquetas e células endoteliais. Adaptado do Sundd, P., et al, 2019.
A anemia falciforme é considerada a patologia genética mais comum no mundo, com incidência de 25% a 40% nos países africanos. A população brasileira é uma mistura de três grupos genéticos, índios americanos, europeus e africanos. O gene da hemoglobina S é muito comum nas Américas e no Brasil, sendo este último mais comum nas regiões sudeste e nordeste. Na África equatorial, onde 40% da população é portadora, a prevalência da doença falciforme atinge 2% a 3% da população. No Brasil, a anemia falciforme atinge entre 0,1% e 0,3% da população negra, e distribui-se heterogeneamente, tendo evidência principalmente nos estados da Bahia, Maranhão e Piauí. A prevalência do traço falciforme é maior nas regiões Norte e Nordeste, entre 6% e 10%, enquanto que nas sul e sudeste é de 2% a 3%, heterozigotos AS alternam de 1 a 3% (4).
De acordo com o protocolo clínico e diretrizes terapêuticas para anemia falciforme do Ministério da Saúde de 2018, 4% da população brasileira tem traço falciforme e entre 25.000 a 50.000 pessoas tem anemia falciforme (HbSS) ou na condição de heterozigose composta ou dupla (SC, SE, SD, Sβ-talassemia). Em 2018 podia variar de 50.000 a 100.000 casos (5).
Estudos têm demonstrado que o genótipo HbSS causa a doença sistêmica mais grave do que os genótipos HbSC e Sβ-talassemia, enquanto os genótipos HbSC e Sβ-talassemia apresentam maior incidência de manifestações oculares, incluindo retinopatia falciforme proliferativa (RFP), podendo levar ao comprometimento visual progressivo em 10 a 20% dos olhos afetados (5).
- Retinopatia falciforme
Diferentes órgãos podem ser acometidos pela anemia falciforme, como pulmões, coração, fígado e rins, e também estruturas oculares (5). Dentre as alterações oftalmológicas causadas pela anemia falciforme, as alterações retinianas são consideradas as mais importantes na doença ocular afetada pela AF (6).
A retinopatia falciforme é causada pelo bloqueio dos vasos sanguíneos na microvasculatura do olho, e dependendo de sua localização e do tecido afetado pode levar a distúrbios visuais, com surgimento de alterações orbitárias, conjuntivais, papilares e retinianas, que podem levar a perda progressiva da visão (7). Pode ser dividida em dois tipos: retinopatia falciforme não proliferativa (RP) e retinopatia falciforme proliferativa (RPF). A forma proliferativa pode ser a principal causa de perda progressiva da visão nesses pacientes, afetando 10% a 20% dos olhos acometidos (8).
A oclusão arteriolar marca o início da patogênese e geralmente ocorre nas áreas periféricas da retina. É considerado um estágio inicial da retinopatia e não significa necessariamente que o paciente evoluirá para o estágio de RFP. Tortuosidade, dilatação e formação de microaneurismas na rede capilar também são observados na fase inicial da RF (8).
Na RF, além das lesões hiperpigmentadas em hemorragias profundas ou sub retinianas (chamadas black sunburst), podem aparecer pequenas hemorragias intra retinianas, possivelmente derivadas da necrose isquêmica da parede do vaso, denominadas salmon-patches (9). Na retinopatia falciforme proliferativa (RFP), os neovasos periféricos da retina assumem uma forma frondosa, chamada de sea fan (8).
A isquemia causada por oclusão repetida de pequenos vasos é considerada a causa mais provável de angiogênese. Apesar de terem a mesma gênese, a retinopatia falciforme proliferativa e não proliferativa têm evoluções diferentes. Goldberg desenvolveu uma classificação de alterações progressivas na retina, divididas em 5 categorias de acordo com a ordem de aparecimento e a gravidade da doença (10).
O estágio I é caracterizado por oclusão arteriolar periférica, causada pelo acúmulo de hemoglobina falciforme, que leva à hipóxia retiniana e rearranjo dos capilares adjacentes, acidose e hiperosmolaridade, podendo levar à trombose. No estágio II, ocorre a anastomose arteriovenosa periférica (hairpin loop) e tem início do brotamento de neovasos que, ao tentar unir a retina vascular com avascular, sofre dilatações levando a distúrbios circulatórios que causam a remodelação vascular que ocorre em resposta à fase I (10).
No estágio III, a neovascularização (sea fan) ocorre possivelmente devido à liberação de fatores angiogênicos na retina isquêmica periférica. Com a neovascularização em sea fan formada, o estágio III é subdividido com base em um ensaio clínico prospectivo de observação versus fotocoagulação retiniana, levando em consideração o tamanho do sea fan, hemorragia, fibrose e visibilidade dos vasos. Os vasos recém-formados são considerados frágeis, imaturos e aderentes ao gel vítreo, o que facilita a ocorrência de hemorragia vítrea, levando ao estágio IV (10). O sangramento repetido leva ao aumento da tensão vítreo retiniana, que pode promover descolamento de retina regmatogênico ou tracional. Quando esse sangramento atinge o eixo visual, desenvolvem-se sintomas e distúrbios visuais. Com eventos hemorrágicos recorrentes, a tração induzida pelo tecido fibroglial e a adesão vítrea à neovascularização aumentam, levando à ruptura, descolamento de retina e perda de visão, estágio final da retinopatia falciforme proliferativa (estágio V). A formação e a tração da membrana resultam de alterações degenerativas no vítreo que superam a neovascularização permeável (10).
A retinopatia falciforme não proliferativa corresponde aos estágios I e II, enquanto a RFP corresponde aos estágios III, IV e V (11). A RFP pode ocorrer em crianças, mas ocorre principalmente na faixa etária entre 15 e 29 anos (12). A regressão espontânea da neovascularização foi relatada em 20% a 60% dos casos (13).
Curiosamente, em homozigotos HbS e heterozigotos compostos HbSC, existe uma relação inversa entre a gravidade da doença sistêmica e a gravidade da retinopatia. Pacientes com anemia falciforme apresentam mais complicações sistêmicas, incluindo múltiplos eventos vaso-oclusivos e danos secundários a órgãos (14). Pacientes heterozigotos têm menos complicações sistêmicas, mas a neovascularização da retina tem maior frequência e início mais precoce (10). A prevalência de RFP é de 32,8% na hemoglobinopatia SC, 14% na Sß-talassemia e 2,6% na SS (15). Em um estudo observaram que a incidência de RFP é cinco vezes mais frequente em pacientes com genótipos HbSC em relação aos pacientes HbSS, e conforme a idade aumenta, se torna mais comum o surgimento dessa complicação (14). Outros estudos relatam que pacientes acima de 35 anos, têm maiores riscos de desenvolver RFP do que pacientes mais jovens, apresentando maiores riscos de perda da visão (estágio III – IV), indicando que a idade é um fator de risco para a RFP (16).
No Brasil, cerca de 20% dos negros são heterozigotos para alfa-talassemia. A heterozigosidade composta com outros tipos de hemoglobina também modula a polimerização da HbS (17). A hemoglobina C (HbC) é a variante estrutural mais comum e juntamente com a HbS (βSβC) leva a um perfil clínico diferente da anemia falciforme (βSβS), resultando em um quadro clínico relativamente benigno, mas com alta prevalência de complicações tromboembólicas, necrose de papilas renais e retinopatia (18). A razão exata pela qual indivíduos com o genótipo βSβC têm mais problemas oculares não é conhecida (19). Acredita-se que os níveis de células falciformes, hematócrito e viscosidade sanguínea tenham um papel importante nessa diferença (20).
Um estudo recente foi realizado por nosso grupo comparando o perfil de expressão gênica obtido pelo sequenciamento de RNA de células endoteliais formadoras de colônia (CEFC) isoladas de sangue de pacientes com genótipos HbSS com e sem retinopatia, e de pacientes HbSC com e sem retinopatia falciforme. Após isolamento, cultura, caracterização das CEFCs, extração de RNA, sequenciamento e análise dos resultados, foi identificada uma lista de genes diferencialmente expressos, destacando-se a expressão significativa do gene ROBO1 em pacientes com genótipo SC (log2FoldChange= 4,3, FDR= 1,35e -11) com RFP, e super expressão do gene RAB38 (log2FoldChange = 3,296, FDR= 2,4e-06) em pacientes com genótipo SS com RFP. Embora pesquisas já tenham vinculado tais genes aos processos de invasão, migração, angiogênese e proliferação celular, mecanismos envolvidos na retinopatia, este estudo tenta vincular tais genes à fisiopatologia da retinopatia falciforme.
- RAB38
O gene Ras-Related Protein Rab-38 (RAB38), está localizado no cromossomo 11, banda q14.2. Este gene está envolvido na regulação da transdução de sinal e processos celulares, incluindo crescimento e diferenciação celular, sendo o principal deles a regulação do transporte e migração de vesículas intracelulares (22). O RAB38 é encontrado em certos organismos, em certas organelas subcelulares e desempenha um papel importante na reprodução celular, endocitose, transdução de sinal e desenvolvimento (23). A subfamília RAB32 consiste em duas proteínas, RAB32 e RAB38, e é conhecida por regular o tráfego de membrana mediada por endossomos de organelas relacionadas ao lisossomo. RAB32 e RAB38 são geneticamente e estruturalmente similares (22).
RAB38 é uma proteína GTPase envolvida na regulação da proliferação celular e sobrevivência celular em tumores. Nos últimos anos, o papel do RAB38 tem sido associado à regulação de diferentes aspectos da progressão tumoral, migração celular, invasão, proliferação, comunicação com células estromais e desenvolvimento de resistência a drogas (23).
Lopes e colaboradores, estudaram os mecanismos de biogênese do melanossoma em células epiteliais pigmentares da retina (RPE). Eles observaram em camundongos que o papel do Rab38 na biogênese do melanossoma nas células do RPE impede a prenilação (a adição de moléculas hidrofóbicas à proteína) e resulta em apenas proteínas citosólicas, tornando-as inativas e instáveis (23). Com a perda da atividade de Rab38, a tirosinase é degradada após sair da rede trans-Golgi (TGN), indicando que Rab38 e Rab32 controlam uma etapa no transporte de enzimas melanogênicas do TGN para melanossomos imaturos.
4. ROBO1
Os ROBOS (Roundabout Guidance Receptor) são uma família de proteínas transmembranares altamente expressas em neurônios do sistema nervoso central (SNC). Eles consistem em cinco domínios de Ig e três domínios de fibronectina tipo III (FN3) em sua região extracelular, seguido por uma região transmembranica e um domínio citoplasmático intracelular (24). Foram identificados quatro receptores ROBOem vertebrados (ROBO 1- 4). Embora tenham domínios citoplasmáticos distintos, seus domínios Ig1 e Ig2 são altamente conservados e acredita-se que sejam responsáveis por mediar a interação ROBO-SLIT (24).
O sistema Slit/Robo é um sistema ligante/receptor conservado que normalmente produz quimiorrepulsão e está envolvido na regulação da orientação e ramificação axonal, além da migração de células neuronais durante o desenvolvimento do SNC. O SLIT2 é um componente pró-angiogênico que induz a formação de novos vasos sanguíneos em tecidos angiogênicos e pode regular positivamente e negativamente a angiogênese ligando-se a ROBO1 e ROBO4, respectivamente (25).
O ROBO1 é um dos quatro genes que codificam o receptor Roundabout (ROBO), e regula vários processos biológicos ligando-se a fendas de proteínas secretadas (Slit 1-3). O seu papel é bem estudado na orientação axonal, e na sinalização Slit-Robo, devido a sua importância na neurogênese, angiogênese, proliferação celular e organogênese (26).
Huang e colegas examinaram os padrões de expressão de Robo1 e Robo4 em vasos da retina usando camundongos C57BL/6J. Eles observaram a expressão de Robo4 em vasos da retina, células ganglionares e camadas de fotorreceptores durante o desenvolvimento da retina. Embora alta expressão de Robo1 tenha sido observada em diferentes estágios do desenvolvimento vascular da retina, eles sugeriram que Robo1 desempenhava um papel importante na neovascularização da retina. Dados tem mostrado níveis de expressão significativamente mais altos de Robo1 em retinas de camundongos com modelo de retinopatia da prematuridade (ROP) induzida por oxigênio (27-29). A ROP é caracterizada por isquemia local e neovascularização pré-retiniana. Rama e colegas observaram evidências genéticas para um importante papel da sinalização Slit2 através de Robo1 e Robo2 na neovascularização ocular patológica e de desenvolvimento (28). Além disso, foi relatado que a angiogênese induzida por VEGF também requer a presença de Robo1 (28).
5. Conclusões
Sendo a retinopatia falciforme, principalmente na sua forma proliferativa, considerada a afecção ocular de maior importância clínica nas doenças falciformes e a maior causa da perda de visão dos indivíduos acometidos pela doença, torna-se necessário a melhor compreensão da doença e de seus mecanismos.
Como na maior parte das complicações nas doenças falciformes, é atribuída à vaso-oclusão o início do comprometimento retiniano. Em contraste com a maior parte das outras comorbidades falciformes, entretanto, a retinopatia está sujeita à regressão espontânea e exibe sua gravidade em aparente relação inversa ao quadro sistêmico.
Tais peculiaridades fazem da retinopatia falciforme um modelo de estudo de grande interesse para a compreensão dos complexos processos intrínsecos envolvidos nessa complicação falciforme. Dessa forma, essa breve revisão demonstra que muito já sabemos sobre as doenças falciformes contudo há muito ainda para investigarmos sobre os mecanismos moleculares e manejos das complicações oculares decorrentes.
AGRADECIMENTOS
Agradeço a Universidade Estadual de Campinas (UNICAMP), ao programa de pós-graduação Clínica médica e a Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) pelo auxílio financeiro.
REFERÊNCIAS
- Kato GJ, Piel FB, Reid CD, Gaston MH, Ohene-Frempong K, Krishnamurti L, Smith WR, Panepinto JA, Weatherall DJ, Costa FF, Vichinsky EP. Sickle cell disease. Nat Rev Dis Primers. 2018 Mar 15; 4:18010. doi: 10.1038/nrdp.2018.10.
- Ingram VM. A specific chemical difference between the globins of normal human and sickle-cell anaemia haemoglobin. Nature. 1956 Oct 13;178(4537):792-4. doi: 10.1038/178792a0. PMID: 13369537.
- Kapoor S, Little JA, Pecker LH. Advances in the Treatment of Sickle Cell Disease. Mayo Clin Proc. 2018 Dec;93(12):1810-1824. doi: 10.1016/j.mayocp.2018.08.001. Epub 2018 Nov 7.
- Soares, Leonardo Ferreira et al. Prevalência de hemoglobinas variantes em comunidades quilombolas no estado do Piauí, Brasil. Ciência & Saúde Coletiva [online]. 2017, v. 22, n. 11, pp. 3773-3780. ISSN 1678-4561. https://doi.org/10.1590/1413-812320172211.04392016.
- Ministério da saúde secretaria de atenção à saúde secretaria de ciência, tecnologia e insumos estratégicos portaria conjunta no 05, de 19 de fevereiro de 2018. Protocolo Clínico e Diretrizes Terapêuticas Da Doença Falciforme. http://portalms.saude.gov.br/protocolos-e-diretrizes,.
- Vilela, Rosana Q. B.; Bandeira, Denise M. and Silva, Maria Alexsandra E..Alterações oculares nas doenças falciformes. Rev. Bras. Hematol. Hemoter. [online]. 2007, vol.29, n.3, pp.285-287. ISSN 1516-8484. Doi: 0.1590/S1516-84842007000300018.
- Abdalla Elsayed MEA, Mura M, Al Dhibi H, Schellini S, Malik R, Kozak I, Schatz P. Sickle cell retinopathy. A focused review. Graefes Arch Clin Exp Ophthalmol. 2019 Jul;257(7):1353-1364. doi: 10.1007/s00417-019-04294-2.
- Lutty, G. A. & McLeod, D. S. Angiogenesis in Sickle Cell Retinopathy. Retin. Choroidal Angiogenes. 389–405 (2008) doi:10.1007/978-1-4020-6780-8_20.
- Bonanomi MTB. Alterações oculares na doença falciforme. In: Anvisa. Manual de diagnóstico e tratamento de doenças falciformes. 1ª edição. 2002.
- Goldberg MF. Classification and pathogenesis of proliferative sickle retinopathy. Am J Ophthalmol. 1971 Mar;71(3):649-65. doi: 10.1016/0002-9394(71)90429-6
- Manfredini, V., Castro, S. & Wagner, S. A fisiopatologia da anemia falciforme. Hemoglobin 4–7 (2007).
- David, Renato Cunha; Moraes Junior, Haroldo Vieira de and Rodrigues, Márcio Penha Morterá.Alterações oculares e eletrorretinográficas na doença falciforme. Arq. Bras. Oftalmol. [online]. 2011, vol.74, n.3, pp.190-194. ISSN 0004-2749. Doi:10.1590/S0004-27492011000300009.
- Balkaran B, Char G, Morris JS, Thomas PW, Serjeant BE, Serjeant GR. Stroke in a cohort of patients with homozygous sickle cell disease. J Pediatr. 1992 Mar;120(3):360-6. doi: 10.1016/s0022-3476(05)80897-2. PMID: 1538280.
- Downes SM, Hambleton IR, Chuang EL, Lois N, Serjeant GR, Bird AC. Incidence and natural history of proliferative sickle cell retinopathy: observations from a cohort study. Ophthalmology. 2005 Nov;112(11):1869-75. doi: 10.1016/j.ophtha.2005.05.026.
- Clarkson JG. The ocular manifestations of sickle-cell disease: a prevalence and natural history study. Trans Am Ophthalmol Soc. 1992;90:481-504. PMID: 1494832; PMCID: PMC1298447.
- Saidkasimova S, Shalchi Z, Mahroo OA, Shunmugam M, Laidlaw DA, Williamson TH, Howard J, Mohamed MD. Risk factors for visual impairment in patients with sickle cell disease in London. Eur J Ophthalmol. 2016 Aug 4;26(5):431-5. doi: 10.5301/ejo.5000767.
- Bianchetti E, Bates SJ, Nguyen TTT, Siegelin MD, Roth KA. RAB38 Facilitates Energy Metabolism and Counteracts Cell Death in Glioblastoma Cells. Cells. 2021 Jun 30;10(7):1643. doi: 10.3390/cells10071643.
- Wang H, Jiang C. RAB38 confers a poor prognosis, associated with malignant progression and subtype preference in glioma. Oncol Rep. 2013 Nov;30(5):2350-6. doi: 10.3892/or.2013.2730. Epub 2013 Sep 10.
- Lopes VS, Wasmeier C, Seabra MC, Futter CE. Melanosome maturation defect in Rab38-deficient retinal pigment epithelium results in instability of immature melanosomes during transient melanogenesis. Mol Biol Cell. 2007 Oct;18(10):3914-27. doi: 10.1091/mbc.e07-03-0268.
- Stewart MW. The expanding role of vascular endothelial growth factor inhibitors in ophthalmology. Mayo Clin Proc. 2012 Jan;87(1):77-88. doi: 10.1016/j.mayocp.2011.10.001.
- Ortiz-Sandoval CG, Hughes SC, Dacks JB, Simmen T. Interaction with the effector dynamin-related protein 1 (Drp1) is an ancient function of Rab32 subfamily proteins. Cell Logist. 2014 Oct 2;4(4):e986399. doi: 10.4161/21592799.2014.986399. PMID: 25767741; PMCID: PMC4355727.
- Coll M, Ariño S, Martínez-Sánchez C, Garcia-Pras E, Gallego J, Moles A, Aguilar-Bravo B, Blaya D, Vallverdú J, Rubio-Tomás T, Lozano JJ, Pose E, Graupera I, Fernández-Vidal A, Pol A, Bataller R, Geng JG, Ginès P, Fernandez M, Sancho-Bru P. Ductular reaction promotes intrahepatic angiogenesis through Slit2-Roundabout 1 signaling. Hepatology. 2022 Feb;75(2):353-368. doi: 10.1002/hep.32140. Epub 2021 Dec 15.
- Scala M, Accogli A, Allegri AME, Tassano E, Severino M, Morana G, Maghnie M, Capra V. Familial ROBO1 deletion associated with ectopic posterior pituitary, duplication of the pituitary stalk and anterior pituitary hypoplasia. J Pediatr Endocrinol Metab. 2019 Jan 28;32(1):95-99. doi: 10.1515/jpem-2018-0272.
- Zhao Y, Yang JY, Thieker DF, Xu Y, Zong C, Boons GJ, Liu J, Woods RJ, Moremen KW, Amster IJ. A Traveling Wave Ion Mobility Spectrometry (TWIMS) Study of the Robo1-Heparan Sulfate Interaction. J Am Soc Mass Spectrom. 2018 Jun;29(6):1153-1165. doi: 10.1007/s13361-018-1903-4. Epub 2018 Mar 8. PMID: 29520710; PMCID: PMC6004239.
- Rama N, Dubrac A, Mathivet T, Ní Chárthaigh RA, Genet G, Cristofaro B, Pibouin-Fragner L, Ma L, Eichmann A, Chédotal A. Slit2 signaling through Robo1 and Robo2 is required for retinal neovascularization. Nat Med. 2015 May;21(5):483-91. doi: 10.1038/nm.3849. Epub 2015 Apr 20.
- Kong Y, Sun B, Han Q, Han S, Wang Y, Chen Y. Slit-miR-218-Robo axis regulates retinal neovascularization. Int J Mol Med. 2016 Apr;37(4):1139-45. doi: 10.3892/ijmm.2016.2511. Epub 2016 Feb 29. Retraction in: Int J Mol Med. 2016 Dec;38(6):1947.
- Duarte DA, Rosales MA, Papadimitriou A, Silva KC, Amancio VH, Mendonça JN, Lopes NP, de Faria JB, de Faria JM. Polyphenol-enriched cocoa protects the diabetic retina from glial reaction through the sirtuin pathway. J Nutr Biochem. 2015 Jan;26(1):64-74. doi: 10.1016/j.jnutbio.2014.09.003. Epub 2014 Oct 2. PMID: 25448608.
- Promsote W, Powell FL, Veean S, Thounaojam M, Markand S, Saul A, Gutsaeva D, Bartoli M, Smith SB, Ganapathy V, Martin PM. Oral Monomethyl Fumarate Therapy Ameliorates Retinopathy in a Humanized Mouse Model of Sickle Cell Disease. Antioxid Redox Signal. 2016 Dec 10;25(17):921-935. doi: 10.1089/ars.2016.6638
- ICite NI of HO of PA (NIH)-. Results for sickle cell disease, retinopathy [Internet]. Available from: https://icite.od.nih.gov/analysis
30. Sundd P, Gladwin MT, Novelli EM. Pathophysiology of Sickle Cell Disease. Annu Rev Pathol. 2019 Jan 24;14:263-292. doi: 10.1146/annurev-pathmechdis-012418-012838. Epub 2018 Oct 17. PMID: 30332562; PMCID: PMC7053558.

{kind=link}