
Capítulo de livro publicado no livro do I CONGEB. Para acessa-lo clique aqui.
DOI: https://doi.org/10.53934/9786585062046-23
Este trabalho foi escrito por:
Marta Marques de Carvalho Lopes *; Thaís Dornas Xavier ; Rejane Alves de Carvalho Monteiro ; Isabela Aurora Rodrigues ; Isabelly Rocha Borges ; Amanda De Lourdes Nunes ; Uyara da Silva Cadar
*Autor Correspondente
E-mail: [email protected]
Resumo: Alterações cromossômicas numéricas e estruturais são, em sua maioria, incompatíveis com a vida, principalmente quando estão presentes em todas as células do organismo. São responsáveis por cerca de 50% de todos os abortos espontâneos ocorridos no primeiro trimestre de gravidez. As translocações balanceadas e não balanceadas, são exemplos de alterações cromossômicas estruturais, sendo as não- balanceadas responsáveis pela formação de cromossomos derivativos. Assim, relatamos um caso de uma criança apresentando um rearranjo cromossômico raro e complexo, envolvendo os cromossomos 5 e 18. Os exames realizados que possibilitaram a detecção de tais rearranjos, foram o Cariótipo de Banda G, o CGH-array eoFISH para a Síndrome de CRI-DU-CHAT/SOTOS. O resultado do exame de Cariótipo de Banda G revelou o resultado 47,XY,t(5;18)(q35;q21.1),+der(18)t(5;18)(q35;q21.1), corroborando com os achados encontrados na análise por CGH-array, que demonstrou um ganho nas regiões 5q35.3 e 18p11.32q21.1, gerando uma trissomia parcial de ambos os cromossomos. Já o exame de FISH apresentou resultado normal. A correlação fenótipo e genótipo constituem-se como uma ferramenta importante no diagnóstico e prognóstico de indivíduos com alterações cromossômicas raras, pois auxiliam no desenvolvimento de novas estratégias terapêuticas que podem proporcionar uma melhor qualidade de vida ao portador.
Palavras-chave: alteração cromossômica rara; Síndrome De Edwards Parcial; cromossomo derivativo; duplicação 5q35
INTRODUÇÃO
A estabilidade numérica e estrutural dos cromossomos em qualquer organismo é considerada um dos principais fatores para o seu desenvolvimento, resultando em um indivíduo físico e psicologicamente normal (1). Qualquer erro nesta dinâmica resultará em alterações cromossômicas que, em sua maioria, são incompatíveis com a vida, principalmente quando acometem todas as células do organismo. Tais alterações, são responsáveis por aproximadamente 50% de todos os abortos espontâneos ocorridos no primeiro trimestre de gestação, acometendo também e 1 a cada 160 nativivos (2).
A translocação balanceada ou recíproca é o tipo de alteração cromossômica estrutural mais comum, onde segmentos cromossômicos de dois ou mais cromossomos são trocados entre si. Portadores dessa alteração constitucional são fenotipicamente normais, entretanto, podem gerar gametas não-balanceados, levando à infertilidade, abortamento espontâneo ou ao nascimento de conceptos anormais (3).
A translocação não-balanceada pode acarretar na formação de cromossomos denominados derivativos, que são aqueles que apresentam o centrômero intacto, ou seja, identificável, mas que sofreram rearranjos intercromossomal e/ou intracromossomal. Assim, pode-se dizer que é um cromossomo estruturalmente alterado que derivou de um cromossomo conhecido. Normalmente, indivíduos que apresentam cromossomos derivativos, herdaram de seus progenitores um dos cromossomos rearranjados devido à presença de translocação recíproca, carreando monossomias/trissomias parciais raras (3).
Neste contexto, apresentamos o relato de uma criança com um rearranjo cromossômico raro, envolvendo os cromossomos 5 e 18, uma translocação recíproca seguida de uma duplicação de um dos cromossomos derivativo, gerando uma trissomia parcial dos mesmos.
MATERIAIS E MÉTODOS
Primeiramente, foi realizada a análise de variações de número de cópias do genoma do paciente, através do exame CGH-array usando CytoScan®750K Array (Affymetrix, Santa Clara, CA, EUA), seguindo os procedimentos descritos pelo fabricante. A análise de dados foi realizada usando o Software Affymetrix® Chromosome Analysis Suite (ChAS). Em seguida, a técnica de FISH – Hibridização in situ Fluorescente foi feita, utilizando a sonda para Síndrome de deleção do cromossomo 5p ou CRI-DU-CHAT/SOTOS PROBE COMBINATION, conforme protocolo padrão (Cytocell Aquarius, Inglaterra, OGT). E, para complementar o diagnóstico do paciente, a análise citogenética de linfócitos do sangue periférico foi realizada usando técnicas padrões de bandamento G (400 bandas).
Todos os exames foram realizados no Departamento de Citogenética e Genética Molecular do Instituto Hermes Pardini S/A. O Termo de Consentimento Livre e Esclarecido (TCLE) autorizando a utilização dos resultados, assim como todas as informações relevantes para a utilização em pesquisa e/ou divulgação científica foi assinado pela responsável do paciente.
Adicionalmente, foi realizada uma revisão bibliográfica nos bancos de dados públicos disponíveis (PubMed, MedLine, DECIPHER, ClinVar) das alterações apresentadas pelo concepto em questão, considerando os fenótipos relacionados e a patogenicidade determinada ou descrita nas alterações semelhantes às do paciente.
RELATO CLÍNICO
O probando é do sexo masculino, atualmente com 5 anos e 3 meses de vida, segundo filho de casal hígido e não consanguíneo. Nasceu a termo, parto cesáreo por pré-eclampsia e com boa vitalidade, porém com características de retardo de crescimento intra-uterino e sindrômico. A mãe, G3P2A1, apresentava ao nascimento 30 anos de idade, nega qualquer exposição a agentes teratógenos durante o período pré-natal. O casal apresenta uma filha hígida com idade atual de 18 anos e não apresenta histórico de má- formações ou deficiência intelectual em ambas as famílias.
O paciente foi submetido à avaliação com o neuropediatra, onde foi observado um atraso do desenvolvimento neuropsicomotor e hipotonia global correspondente para a idade, principalmente o comprometimento do intelecto/cognição e linguagem expressiva aliada à alteração motora. Não possui cardiopatia congênita, apresenta dismorfias faciais, criptorquidia à esquerda, dificuldade de deglutição, sialorreia excessiva e sem controle esfincteriano. No exame de Tomografia computadorizada foi evidenciado um aumento da fossa posterior.
O resultado do exame de CGH-array revelou um ganho de aproximadamente 47 Mb no cromossomo 18 na região p11.32q21.1, envolvendo cerca de 258 genes e de 3,4 Mb no cromossomo 5 na região q35.3, compreendendo cerca de 64 genes, resultando em uma trissomia parcial dos cromossomos 5 e 18. Ambas as variações foram classificadas como patogênicas de acordo com os bancos de dados (DECIPHER e ClinVar). Essas alterações compreendem alguns genes com funções conhecidas que poderiam estar modulando para o fenótipo do paciente. Além de duas CNVs (variações no número de cópias), encontradas na população em geral, sem associações até o momento, a fenótipos anormais.
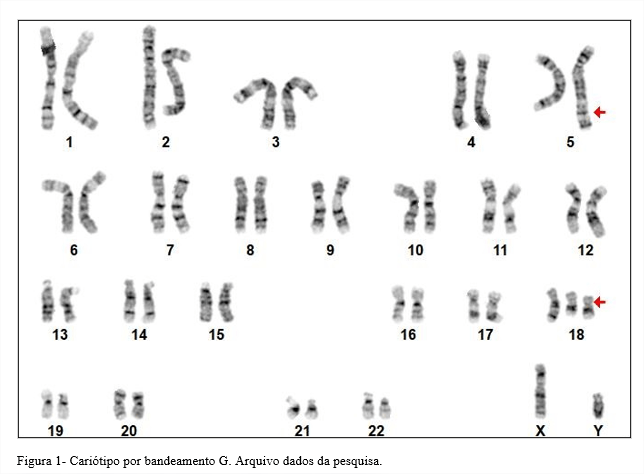
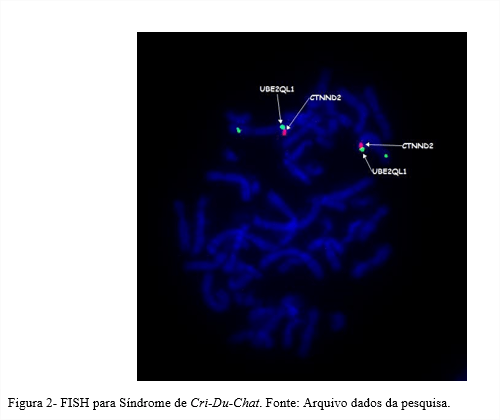
No exame de Cariótipo de Banda G, também foi identificada a trissomia parcial dos cromossomos 5 e 18 (Figura 1). A pesquisa para Síndrome de Cri-Du-Chat através da técnica de FISH (Figura 2) foi negativa.
Na tabela 1, encontram-se os resultados de todos os exames realizados para diagnóstico do paciente.
Tabela 1- Resultados dos exames realizados no Instituto Hermes Pardini S/A.
| Exame | Resultado |
| CGH-array | -arr [hg19] 18p11.32q21.1 (136, 227-47, 327,624)x3 -arr [hg19] 5q35.3 (177,259, 401-180, 715, 096)x3 -2 CNVs presentes em indivíduos da população em geral |
| Cariótipo Banda G | 47,XY,t(5;18)(q35;q21.1),+der(18)t(5;18)(q35;q21.1) |
| FISH Cri-Du-Chat | ish (CTNND2,UBE2QL1)x2 |
Legenda: CGH-array: hibridização genômica comparativa por microarranjos; CNVs: Variações do número de cópia; FISH: Hibridização in situ por fluorescência. Fonte: Arquivo dados da pesquisa.
No momento, não há ainda tratamento específico, somente as terapias de reabilitação. Devido à pandemia causada pelo Coronavírus-Covid-19 a criança tem apresentado uma regressão. As terapias de reabilitação incluem terapia ocupacional e fonoaudiologia com certificação em BOBATH OU PADOVAN, fisioterapia motora com certificação em PEDIASUIT e aquática com especialização em ABA, divididas durante a semana.
DISCUSSÃO
Com os avanços das técnicas citogenômicas, novos loci estão sendo relacionados e determinados para clínicas específicas. Consequentemente, novos genes, localizados nesses novos loci cromossômicos, vêm sendo apontados como candidatos responsáveis pelas alterações clínicas apresentadas pelo paciente ou por determinada síndrome (3,4).
O resultado exame CGH-array permitiu identificar cópias extras de vários genes localizados em 5q35.3 e 18p11.32q21.1, variantes consideradas patogênicas (5). Além de duas CNVs descritas em indivíduos da população em geral. Relacionar alterações no padrão de expressão gênica com as variações no número de cópias no genoma constitui-se uma tarefa difícil, visto que as mesmas estão presentes tanto em indivíduos fenotipicamente normais, como em indivíduos fenotipicamente afetados. Estima-se que cerca de 12% de todo o genoma humano é alvo de variações no número de cópias, conferindo uma importante contribuição para a variabilidade genética presente na espécie humana (5,6).
Através dos achados encontrados no Cariótipo de Banda G, foi possível confirmar e definir qual tipo de alteração cromossômica estrutural o paciente apresentava, visto que a técnica de CGH-array possibilita detectar perdas ou ganhos, não detectando assim translocações balanceadas. O resultado demonstrou uma translocação balanceada entre os cromossomos 5 e 18, seguida de uma duplicação do cromossomo 18 derivativo, confirmando o ganho das cópias extras de ambos os cromossomos (7). A criança apresenta uma irmã filha dos mesmos pais sem nenhuma anormalidade fenotípica. Não foi possível realizar o cariótipo dos pais e da irmã em tempo hábil para descrever no estudo, que seria de grande valia na elucidação diagnóstica e aconselhamento genético, pois as mesmas poderiam ser herdadas.
A duplicação em 5q35.3 afeta a região terminal do braço longo do cromossomo 5, estando relacionada a uma ampla gama de fenótipos clínicos, porém, até o momento, sem associação a uma síndrome genética especifica. Nesta região, estão mapeados 64 genes, onde a dosagem extra cursa com baixa estatura, dismorfias craniofaciais, microcefalia, deficiência intelectual, retardo na linguagem, atraso motor, alterações no sistema nervoso central, entre outras (8). Tais alterações fenotípicas já descritas em indivíduos com duplicação de 5q35.3 estão de acordo com o fenótipo do paciente alvo do estudo como já descrito, considerando que o ele apresenta atraso do desenvolvimento neuropsicomotor, comprometimento intelectual e de cognição, dismorfias faciais e alterações motoras.
O ganho de 18p11.32q21.1 de aproximadamente 47 Mb relaciona-se com a Síndrome da Trissomia do Cromossomo 18, também conhecida como Síndrome de Edwards. As características apresentadas pelos portadores desta síndrome são muito variáveis e estão diretamente relacionadas com o tamanho do material genético extra (9). A trissomia completa ou total do cromossomo 18 é a forma mais comum na população, representando cerca de 94% dos casos. Trissomias parciais ou em mosaico exibem fenótipos mais brandos. As características típicas incluem deficiência intelectual grave, baixo peso ao nascimento, anormalidades craniofaciais, malformações de órgãos interno, sobreposição dos dedos das mãos e pés, entre outras (9). Todas as dismorfias e alterações neuropsicomotores observadas no paciente também corroboram com as descritas na literatura. Muitas características fenotípicas presente na Síndrome de Edwards sobrepõem as descritas na duplicação de 5q35.3, não sendo possível distinguir até o momento, qual região extra estaria mais relacionada a clínica da criança.
Deste modo, estudos citogenéticos clássicos e moleculares são de suma importância no diagnóstico de portadores de malformações e atraso no desenvolvimento, pois possibilitam a detecção de alterações cromossômicas numéricas e estruturais que possivelmente estariam contribuindo para o fenótipo apresentado.
CONCLUSÕES
Relatos de pacientes portadores de alterações genéticas raras juntamente com as características clínicas apresentadas são de suma importância, pois se constitui como uma ferramenta útil para a correlação genótipo e fenótipo, contribuindo assim para uma melhor compreensão da complexidade da doença, possibilitando também melhores estratégias terapêuticas e medicamentosas, a fim de proporcionar uma melhor qualidade de vida aos indivíduos afetados.
AGRADECIMENTO
Ao Instituto Hermes Pardini S/A pelo apoio e financiamento.
REFERÊNCIAS
1. Nussbaum RL, Mcinnes RR, Willard HF. Genética Médica. Rio de Janeiro: Elsevier; 2008.
2. Blue NR, Page JM, Silver RM. Genetic abnormalities and pregnancy loss. Semin Perinatol. 2019; 43: 66–73.
3. Maluf, SW, Riegel M, et al. Citogenética Humana. Porto Alegre: Artmed; 2011.
4. Levy B, Wapner R. Prenatal diagnosis by chromosomal microarray analysis. Fertil Steril. 2018; 109:201–12.
5. de Leeuw N, et al. Diagnostic interpretation of array data using public databases and internet sources. Hum Mutat. 2012; 33: 930-40.
6. Girirajan S, Eichler EE. Phenotypic variability and genetic susceptibility to genomic disorders. Hum Mol Genet. 2010; 19: R176-R187.
7. Hanemaaijer NM, et al. Practical guidelines for interpreting copy number gains detected by highresolution array in routine diagnostics. Eur J Hum Genet. 2012; 202: 161-165.
8. Yue, Fagui et al. “Prenatal diagnosis of a 5q35.3 microduplication involving part of the ADAMTS2 locus: a likely benign variant without apparent phenotypic abnormality: Case series.” Medicine. 2019. 98:49.
9. Cereda A, Carey JC. The trisomy 18 syndrome. Orphanet J Rare Dis. 2012 Oct 23; 7:81.

{kind=link}