
Capítulo de livro publicado no livro do I CONGEB. Para acessa-lo clique aqui.
DOI: https://doi.org/10.53934/9786585062046-12
Este trabalho foi escrito por:
Rahisa Helena da Silva*; Manassés Daniel da Silva ; Antonio Félix da Costa; Éderson Akio Kido
¹Laboratório de Genética Molecular de Plantas – UFPE
²Instituto Agronômico de Pernambuco
*Autor correspondente (Rahisa Helena da Silva) – Email: [email protected]
Resumo: Desde a descoberta da estrutura do DNA em 1953 por Watson e Crick, através dos dados dos estudos de cristalografia de raios X, obtidos por Rosalind Franklin e Maurice Wilkins, muito se avançou. O rápido crescimento das tecnologias de sequenciamento (DNA e RNA) proporcionou o surgimento das ômicas. As ômicas correspondem a um campo da Biologia Molecular que geram dados capazes de fornecer visões globais sobre a informação genética contida nos organismos. A genômica tem como objeto de estudo o genoma de um dado organismo de interesse, que inclui regiões codificantes e não-codificantes. Desde a escolha da plataforma de sequenciamento mais adequada, a obtenção de um genoma de referência é um processo que abrange várias etapas. Ainda, considerando a complexidade dos genomas vegetais devido a poliploidia e regiões repetitivas muito extensas, o conhecimento sobre as etapas relativas é de extrema importância. Sendo assim, a disponibilidade de um genoma de referência permite a caracterização dos seus componentes, proporcionando o entendimento de mecanismos moleculares associados a processos de desenvolvimento e adaptação. Portanto, o objetivo do capítulo é apresentar os principais aspectos relacionados com a obtenção de um genoma vegetal, passando pelas etapas a serem seguidas e as ferramentas aplicadas.
Palavras–chave: SEQUENCIAMENTO; BIOINFORMÁTICA; GENOMA; ÔMICAS
INTRODUÇÃO
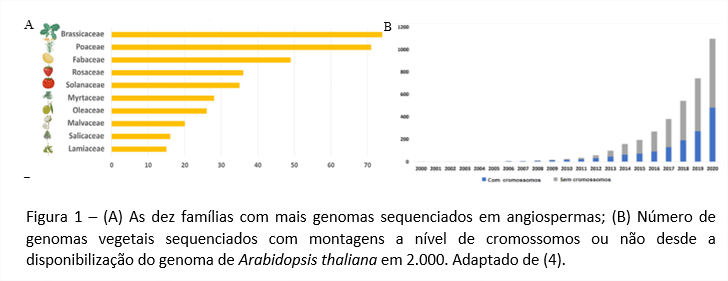
As ciências ômicas são amplamente aplicadas para entender a organização das bases das funções celulares. As ômicas consistem numa abordagem integrativa que reúnem dados de experimentos em larga escala, que incluem a genômica, transcriptômica, proteômica e metabolômica (1). Neste capítulo, vamos abordar aspectos relacionados ao estudo da genômica em plantas, além das ferramentas aplicadas e seu papel no entendimento da biologia desses organismos. O aumento da disponibilidade de genomas vegetais está associado ao avanço nas tecnologias de sequenciamento e da bioinformática, proporcionando análises cada vez mais detalhadas de caracteres de importância econômica, tais como resistência a estresses bióticos e abióticos (2). Em plantas, o primeiro genoma de referência foi obtido de Arabidopsis thaliana (The Arabidopsis Genome Initiative, 2000). Cinco anos depois, o genoma de arroz foi completado utilizando cromossomos artificiais de bactérias (BACs – Bacterial Artificial Chromosomes) e sequenciamento de Sanger (3). Ao final de 2020, 1.031 genomas de referência ou drafts tinham sido disponibilizados para 788 espécies de plantas (4) (Figura 1A).
O custo do sequenciamento foi drasticamente reduzido pelas tecnologias de sequenciamento de nova geração (NGS – Next-generation sequencing), permitindo um aumento explosivo na quantidade de genomas obtidos (4). Dentre algumas das plataformas NGS, podemos citar: 454 (Roche), SOLiD (Life Technologies), HiSeq (Illumina), MiSeq (Illumina) e Ion Torrent (Life Technologies). Entretanto, apesar de as técnicas NGS terem proporcionado maior quantidade de informação disponível, a complexidade da montagem devido aos fragmentos short reads gerados foi responsável pela menor qualidade da informação obtida (5). Esse é um aspecto importante quando se trata de genomas vegetais, que apresentam regiões repetitivas muito extensas, grande tamanho dos genomas e poliploidia, que constituem grandes desafios para a etapa de montagem (6). Sendo assim, o nível de cromossomos era mais difícil de ser alcançado para essas montagens. Logo, o desenvolvimento das tecnologias de sequenciamento de terceira geração, caracterizadas por fragmentos long reads, foi essencial para a melhoria da acurácia e contiguidade dos genomas e serem disponibilizados (6) (Figura 1B).
As tecnologias de sequenciamento de terceira geração podem ser divididas em duas abordagens: o sequenciamento long-read e a long-range scaffolding (7). Dentre as técnicas de sequenciamento long read, a que utiliza o método Single Molecule Real-Time (SMRT) provido pela Pacific Biosciences’ ou PacBio, é a mais utilizada, com tamanho médio de reads de 20 Kb. A tecnologia Synthetic Long-Reads (SLR), desenvolvida pela Illumina apresenta média de reads de até 10 Kb. Um outro exemplo é o MinION desenvolvido pela Oxford Nanopore, que pode gerar reads de até 12 Kb (8). A técnica de captura da conformação dos cromossomos (Hi-C) representa uma tecnologia de long-range scaffolding, esse protocolo é provido pela Dovetail Genomics (www.dovetails.genomics.com) e auxilia bastante na obtenção de montagens em escala cromossômica – (9). Logo, o maior requisito para estudos em genômica é a disponibilidade de um genoma de referência relativo à espécie a ser analisada. Nos tópicos a seguir serão descritas as etapas referentes à obtenção de um genoma de referência.
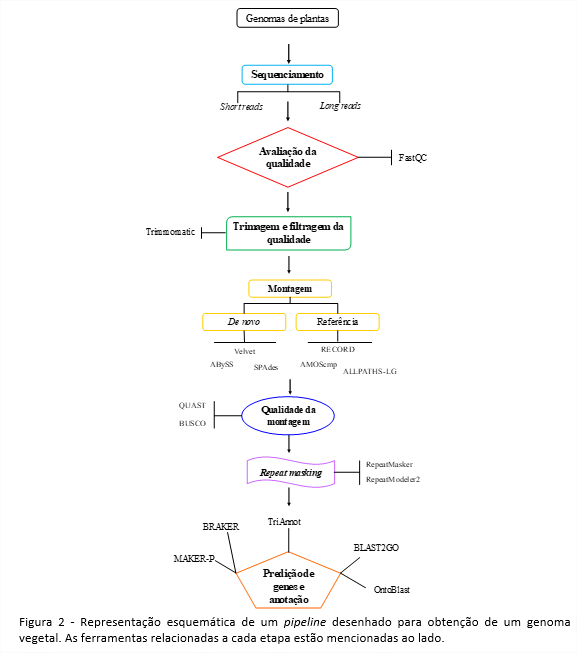
CONTROLE DA QUALIDADE DO SEQUENCIAMENTO
A primeira etapa na análise de dados de sequenciamento de alto rendimento é o controle da qualidade. Nessa etapa, é avaliado um conjunto de estatísticas em um arquivo de reads, que abrange a contagem de frequência relativa de nucleotídeos em cada posição em um conjunto de leituras, para detectar desvios das frequências esperadas, considerando a distribuição de Phred (10). Esse controle de qualidade também mede a frequência de artefatos que não fazem parte da amostra biológica de interesse, como contaminantes e adaptadores. O início dessa etapa ocorre pela conversão dos dados em formato .SRA (Sequencing Read Archive) para o formato .FASTQ por meio da ferramenta SRA toolkit (http://www.ncbi.nlm.nih.gov/Traces/sra/). Para checagem da qualidade propriamente dita, a ferramenta FastQC é a mais amplamente empregada e permite visualização rápida dos dados e dos possíveis pontos problemáticos dentro do sequenciamento (11). Após o processo de checagem, esses dados devem ser pré-processados para remoção de sequências de baixa qualidade e de artefatos do sequenciamento que podem facilmente comprometer análises posteriores. Essa etapa é conhecida como trimagem. Uma das principais ferramentas empregadas para esse objetivo é o Trimmomatic (12). As principais etapas realizadas são: a trimagem ou remoção de sequências técnicas (ex.: adaptadores e primers) e a filtragem da qualidade, que avalia o score de qualidade de cada base sequenciada (frequentemente valor de Phred maior ou igual a 25 ou 30) (Figura 2).
MONTAGEM E AVALIAÇÃO DA QUALIDADE
A montagem de genomas abrange duas abordagens básicas. A primeira é a abordagem de referência, que consiste na utilização de sequências já montadas de organismos taxonomicamente relacionados. A segunda é a abordagem conhecida como de novo, que monta sequências de organismos sem a necessidade de sequências anteriormente sequenciadas (13). O emprego de uma das duas abordagens não exclui a outra, de forma que até em casos onde há disponibilidade de genomas de referência, regiões que variam muito no genoma necessitam da abordagem de novo (5). A abordagem de referência é bastante utilizada, por exemplo, no re-sequenciamento de genomas para corrigir erros de montagem ou estender contigs já existentes, bem como para identificação de variantes (5). Na abordagem de novo, a sequência do genoma é construída através da sobreposição das reads sequenciadas, comumente utilizando algoritmos baseados em gráficos. As short reads costumam dificultar essa abordagem.
Os principais algoritmos de montagem são: OLC (overlap-layout consensus) e o gráfico de Bruijn (DBG) (4). Uma gama de softwares montadores já foram desenvolvidos com o objetivo de obter a sequência completa de genomas. Dentre os montadores que seguem a abordagem por referência estão: RECORD (14), AMOScmp (15), ALLPATHS-LG (16) e CAP3 (17). Dentre os montadores para montagem de novo podemos citar o Velvet (18), ABySS (Assembly By Short Sequences) (19), SPAdes (20) e MaSuRCA (Maryland Super-Read Celera Assembler) (21). Há também montadores especificamente construídos para lidar com o processamento de long reads que incluem: FALCON (22), NECAT (22), Mecat2 (23) e Canu (24), Raven (25) dentre outros (Figura 2).
Após a montagem é essencial entender a qualidade das sequências obtidas, se a acurácia está baixa ou se existem erros de montagem. Esse processo está relacionado com a obtenção de várias métricas que indicam a qualidade da montagem (26). Dentre elas, a mais conhecida é o N50, que se refere ao tamanho do menor contig/scaffold em que metade da montagem está representada. Uma variante do N50 é NG50, que constitui o tamanho do menor contig/scaffold que se encontra na metade de um genoma de referência. Outra métrica muito utilizada é o L50, que representa quantos contigs/scaffolds foram necessários para alcançar metade do sequenciamento. Dentre os softwares desenhados para essa etapa pode-se citar o QUAST (Quality Assessment Tool for Genome Assemblies) (27), BUSCO (Benchmarking Universal Single-Copy Orthologs) (28) e GenomeQC (29) (Figura 2).
REPEAT MASKING
A natureza altamente repetitiva dos genomas vegetais requer que as regiões de repetição sejam mascaradas. Esse processo é chamado de Repeat masking. Resumidamente, as sequências repetitivas abrangem três classes: (1) repetições locais, que incluem repetições simples e em tandem; (2) famílias de repetições dispersas, que compreendem os elementos transponíveis; (3) duplicações segmentais (30). Ocorre que a presença de ORFs (Open Reading Frames) dentro de transposons (ex.: transposases e transcriptases reversas) pode confundir a etapa de predição de genes na montagem (30). As estratégias aplicadas para identificação de elementos repetitivos abrangem duas grandes categorias: (1) detecção de novo e (2) detecção por similaridade.
Basicamente, esse processo ocorre pela busca de características estruturais, tais como domínios associados a transposons, utilizando bancos de dados de elementos repetitivos conhecidos, como o RepBase (https://www.girinst.org/repbase/) (31) e o TIGR (https://www.hsls.pitt.edu/) (32). As ferramentas RepeatMasker (https://www.repeatmasker.org/) (33) e RepeatModeler2 (http://www.repeatmasker.org/RepeatModeler/) (34) são amplamente empregadas para esse objetivo. RepeatMasker emprega a abordagem de similaridade e utiliza as sequências disponíveis em bibliotecas (ex.: RepBase) para identificar as famílias de repetições em determinado genoma. RepeatMasker é essencial considerando famílias de repetições já descritas, no entanto a descoberta de novas famílias de elementos repetitivos espécie-específicas requer ferramentas como o RepeatModeler2 que utiliza algoritmos para detecção de novo dessas repetições tais como RECON (29) e RepeatScout (35) (Figura 2).
PREDIÇÃO DE GENES E ANOTAÇÃO
A predição de genes é a etapa mais importante da anotação de um genoma. Dois tipos de abordagem estão associados a esse processo: (1) ab initio e (2) baseada em similaridade. Na predição ab initio, as ferramentas são desenhadas para identificar elementos característicos da estrutura dos genes (ex.: TATA-box, sítios de splicing, éxons, íntrons, sítios de iniciação e terminação da tradução). Os preditores ab initio utilizam frequentemente Hidden Markov Model (HMM) e programação dinâmica (36). Uma grande variedade de softwares preditores está disponível, dentre eles pode-se citar FGENESH (37), GeneMark.hmm (38), GENESCAN (39) que apresentam boa performance em plantas (40). A anotação de genomas vegetais também conta com alguns pipelines automáticos como TriAnnot (41), Seqping (42), BRAKER (43), MAKER-P (44) e AUGUSTUS (45). O processo de anotação frequentemente utiliza a estratégia de vocabulários controlados. Gene Ontology (GO – http://www.geneontology.org) é o esquema mais extensivamente utilizado para descrever a função de produtos gênicos (46) (Figura 2).
Essa descrição é feita com base em três ontologias independentes, denominadas: processo biológico, função molecular e componente celular (46). Dentre as ferramentas e recursos que foram desenvolvidos para anotação funcional e mineração de novas sequências através de vocabulários controlados pode-se citar: BLAST2GO (www.blast2go.org) (47), InterProScan (https://www.ebi.ac.uk/interpro) (48), OntoBlast (http://functionalgenomics.de/ontogate) (49), AmiGO (http://amigo.geneontology.org/) (50), GoBlet ( http://goblet.molgen.mpg.de.) (51) dentre outros.
ANÁLISES DOWNSTREAM
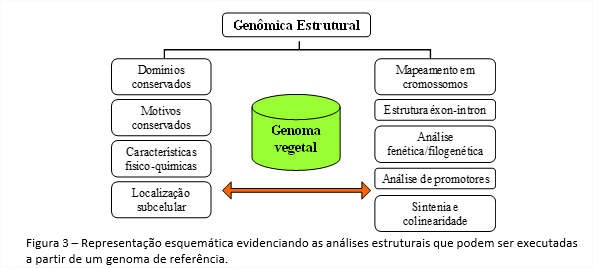
A etapa conhecida como genômica estrutural é o ponto de partida do processo que visa compreender como as estruturas que compõem o genoma estão organizadas e como contribuem para o desempenho de suas funções. A partir da obtenção do genoma de referência, uma variedade de ferramentas pode ser empregada em análises tais como identificação de genes e de seus ortólogos, avaliação da estrutura gênica, análise filogenética/fenética, localização cromossômica, identificação de elementos cis regulatórios em promotores, dentre outros (Figura 3). Dentre algumas das ferramentas empregadas nessa etapa, pode-se citar:
– TBtools (Toolkit for Biologists integrating various biological data-handling tools) abrange mais de 130 funções, que incluem manipulação de arquivos GTF/GFF3, conversão do formato de arquivos, representação gráfica de motivos, domínios e estruturas éxon-íntron, mapeamento de genes em cromossomos, visualização de sintenia, edição e anotação de árvores filogenéticas, visualização de dados de expressão dentre outras (52).
– GSDS (Gene Structure Display Server) (http://gsds.gao-lab.org/Gsds_about.php) é um recurso web amplamente empregado para visualização de estruturas éxon-íntron (53).
– PlantPAN Plant Promoter Analysis Navigator (http://plantpan2.itps.ncku.edu.tw/) é um recurso web para detecção de sítios de ligação de fatores de transcrição (TFBSs), fatores de transcrição correspondentes e outros elementos regulatórios a partir de sequências de promotores (54).
– As ferramentas OrthoFinder (55), OrthoMCL (56) e Inparanoid8 (https://inparanoid.sbc.su.se/cgi-bin/index.cgi) (57) são amplamente empregadas para detecção de ortólogos.
– McScanX é utilizado para detecção de eventos de duplicação (segmental, dispersa, proximal e tandem), estimar número de eventos WGD (whole-genome duplication), detecção de ortólogos colineares e análise de expansão de famílias gênicas (58).
– Os softwares MEGA (Molecular Evolutionary Genetics Analysis) (59) e Clustal (60) são ferramentas extensivamente utilizadas para realizar alinhamentos múltiplos e construção de árvores fenéticas/filogenéticas.
– Os recursos online Evolview (https://www.evolgenius.info/evolview) (61) e iTOL (https://itol.embl.de/) (62) são empregados para visualização, anotação e edição de árvores.
Existem também as ferramentas voltadas especificamente para análise da estrutura de proteínas, são estas:
– SMART Simple Modular Architecture Research Tool (http://smart.embl-heidelberg.de/) é um recurso web para identificação e anotação de domínios de proteínas e visualização de suas arquiteturas (63).
– CDD Conserved Domain Database é um banco de dados localizado na entrez protein do NCBI, é amplamente utilizado para identificação de domínios conservados (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) (64).
– PFAM (http://pfam.xfam.org/) é um banco de dados de famílias proteicas que abriga perfis HMM para identificação de mais de 19.000 famílias (65).
– Jalview é um software para edição, análise e anotação de alinhamentos múltiplos, além de predição de estruturas secundárias (66).
– MEME suite é um servidor web para detecção, análise e visualização de assinaturas de sequência (motivos) presentes em um dado input (67).
– As ferramentas ExPasy (https://web.expasy.org/protparam/) (68) e JVirGel (http://www.jvirgel.de/) (69) computam parâmetros físico-químicos a partir das sequências peptídicas, tais como composição de aminoácidos, ponto isoelétrico e peso molecular.
– As ferramentas CELLO (http://cello.life.nctu.edu.tw/) (70), Wolf Psort (https://wolfpsort.hgc.jp/) (71) e LOCALIZER (https://localizer.csiro.au/) (72) são recursos empregados para identificação das localizações subcelulares de proteínas.
CONCLUSÕES
A obtenção de um genoma de referência de alta qualidade confere benefícios em análises genéticas posteriores de caracterização dos elementos que compõem esse genoma bem como, na tomada de decisões em programas de melhoramento. Dentre essas vantagens, a identificação de genes e sua associação com mecanismos moleculares que atuam na variação de traços de interesse econômico, tais como aqueles associados com maior produtividade, qualidade de frutos ou tolerância a estresses bióticos e abióticos. Os dados de estudos genômicos são de fundamental importância pois permitem o melhor entendimento da base genética da diversidade em plantas, além de facilitar a introdução de traços de interesse mediante técnicas de transformação genética para obtenção de variedades superiores.
REFERÊNCIAS
- Fukushima A, Kusano M, Redestig H, Arita M, Saito K. Integrated omics approaches in plant systems biology 2009.
- Neale DB, Mart PJ, Torre ARD La, Montanari S, Wei X. Novel Insights into Tree Biology and Genome Evolution as Revealed Through Genomics 2017.
- Project, International Rice Genome Sequencing. “The map-based sequence of the rice genome.” Nature 436.7052 (2005): 793-800.
- Sun Y, Shang L, Zhu Q, Fan L. Twenty years of plant genome sequencing: achievements and challenges. Trends Plant Sci 2021;27:391–401.
- Kyriakidou M, Tai HH, Anglin NL, Ellis D, Strömvik M V. Current Strategies of Polyploid Plant Genome Sequence Assembly 2018;9:1–15.
- Kersey PJ. ScienceDirect Plant genome sequences: past, present , future 2019:1–8.
- Jiao W, Schneeberger K. ScienceDirect The impact of third generation genomic technologies on plant genome assembly. Curr Opin Plant Biol 2017;36:64–70.
- Schmidt MH, Vogel A, Denton AK, Istace B, Wormit A, Geest H Van De, et al. De Novo Assembly of a New Solanum pennellii Accession Using Nanopore Sequencing 2017;29:2336–48.
- Lieberman-aiden E. Comprehensive Mapping of Long-Range 2012;289.
- Phred U, Ewing B, Green P. Base-Calling of Automated Sequencer Traces 1998:186–94.
- Andrews S: FastQC: a quality control tool for high throughput sequence data.2010.
- Bolger ME, Weisshaar B, Scholz U, Stein N, Usadel B, Mayer KFX. Plant genome sequencing – applications for crop improvement. Curr Opin Biotechnol 2014;26:31–7.
- Pop M. Genome assembly reborn: recent computational challenges 2009;10:354–66.
- Buza K, Wilczynski B, Dojer N. RECORD : Reference-Assisted Genome Assembly for Closely Related Genomes 2015;2015.
- Pop M, Phillippy A, Delcher AL, Salzberg SL. Comparative genome assembly 2004;5:237–48.
- Gnerre S, Maccallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data 2011;108:1513–8.
- Huang X, Madan A. CAP3 : A DNA Sequence Assembly Program 1999:868–77.
- Zerbino DR, Birney E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs 2008:821–9.
- Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJM. ABySS: A parallel assembler for short read sequence data 2009:1117–23.
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. and Its Applications to Single-Cell Sequencing 2012;19:455–77.
- Zimin, AV, Marçais G, Puiu D, Roberts M, Salzberg SL, Yorke, JA (2013). The MaSuRCA genome assembler. Bioinformatics, 29(21), 2669-2677.
- Chin C, Peluso P, Sedlazeck FJ, Nattestad M, Concepcion GT, Clum A, et al. Phased diploid genome assembly with single-molecule real-time sequencing 2016;13.
- Chen Y, Nie F, Xie S, Zheng Y, Bray T, Dai Q. Fast and accurate assembly of Nanopore reads via progressive error correction and adaptive read selection 2020.
- Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. Canu : scalable and accurate long-read assembly via adaptive k -mer weighting and repeat separation 2017:722–36.
- Vaser R, Mile Š. Raven: a de novo genome assembler for long reads 2021:2–10.
- Wajid B, Sohail MU, Ekti AR, Serpedin E. The A, C, G, and T of Genome Assembly 2016;2016.
- Gurevich A, Saveliev V, Vyahhi N, Tesler G. BIOINFORMATICS APPLICATIONS NOTE Genome analysis QUAST: quality assessment tool for genome assemblies 2013;29:1072–5.
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva E V, Zdobnov EM. BUSCO : assessing genome assembly and annotation completeness with single-copy orthologs 2015;31:3210–2.
- Manchanda N, Ii JLP, Woodhouse MR, Seetharam AS, Lawrence-dill CJ, Andorf CM, et al. GenomeQC: a quality assessment tool for genome assemblies and gene structure annotations 2020:1–9.
- Bao Z, Eddy SR. Automated De Novo Identification of Repeat Sequence Families in Sequenced Genomes 2002:1269–76.
- Bao W, Kojima KK, Kohany O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob DNA 2015:4–9.
- Ouyang S, Buell CR. The TIGR Plant Repeat Databases: a collective resource for the identification of repetitive sequences in plants 2004;32:360–3.
- Chen RAFA, Green P. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences 2004:1–14.
- Flynn JM, Hubley R, Goubert C, Rosen J, Clark AG. RepeatModeler2 for automated genomic discovery of transposable element families 2020;117:9451–7.
- Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes 2005;21:351–8.
- Ouyang S, Thibaud-nissen F, Childs KL, Zhu W, Buell CR. Chapter 14 Plant Genome Annotation Methods n.d.;513:263–82.
- Salamov AA, Solovyev V V. Ab initio Gene Finding in Drosophila Genomic DNA 2000:516–22.
- Lomsadze A, Ter-Hovhannisyan V, Chernoff YO, Borodovsky M. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res 2005;33:6494–506.
- Burge C, Karlin S. Prediction of Complete Gene Structures in Human Genomic DNA 1997:78–94.
- Yao H, Guo L, Fu Y, Borsuk LA, Wen T, Skibbe S, et al. Evaluation of five ab initio gene prediction programs for the discovery of maize genes 2005:445–60.
- Leroy P, Guilhot N, Sakai H, Bernard A, Choulet F, Theil S. TriAnnot: a versatile and high performance pipeline for the automated annotation of plant genomes 2012;3:1–14.
- Chan K, Rosli R, Tatarinova T V, Hogan M, Firdaus-raih M, Low EL. Seqping : gene prediction pipeline for plant genomes using self-training gene models and transcriptomic data. BMC Bioinformatics 2017;18:1–7.
- Hoff KJ, Lange S, Lomsadze A, Borodovsky M, Stanke M. BRAKER1: Unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 2016;32:767–9.
- Campbell MS, Law M, Holt C, Stein JC, Moghe GD, et al. MAKER-P: A Tool Kit for the Rapid Creation, Management, and Quality Control of Plant 2014;164:513–24.
- Stanke M, Morgenstern B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res 2005;33:465–7.
- Gene T, Consortium O. Gene Ontology: tool for the 2000;25:25–9.
- Conesa A, Stefan G. Blast2GO: A Comprehensive Suite for Functional Analysis in 2008;2008.
- Zdobnov EM, Apweiler R. signature-recognition methods in InterPro 2001;17:847–8.
- Genetics M. OntoBlast function: from sequence similarities directly to potential functional annotations by ontology terms 2003;31:3799–803.
- Carbon S, Ireland A, Mungall CJ, Shu S, Marshall B, Lewis S, et al. AmiGO: online access to ontology and annotation data 2009;25:288–9.
- Groth D, Lehrach H, Hennig S. GOblet: a platform for Gene Ontology annotation of anonymous sequence data 2004;32:313–7.
- Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, et al. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol Plant 2020;13:1194–202.
- Hu B, Jin J, Guo A, Zhang H, Luo J. Genome analysis GSDS 2.0: an upgraded gene feature visualization server 2015;31:1296–7.
- Chow C, Zheng H, Wu N, Chien C, Huang H, Lee T, et al. PlantPAN 2 .0: an update of plant promoter analysis navigator for reconstructing transcriptional regulatory 2016;44:1154–60.
- Emms DM, Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics 2019:1–14.
- Li L, Jr CJS, Roos DS. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes 2003:2178–89.
- Sonnhammer ELL, Ostlund G. InParanoid 8: orthology analysis between 273 proteomes, mostly eukaryotic 2015;43:234–9.
- Wang Y, Tang H, Debarry JD, Tan X, Li J, Wang X, et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity 2012;40:1–14.
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) Software Version 4.0 n.d.:0–3.
- Larkin MA, Blackshields G, Brown NP, Chenna R, Mcgettigan PA, Mcwilliam H, et al. Clustal W and Clustal X version 2 .0 2007;23:2947–8.
- Subramanian B, Gao S, Lercher MJ, Hu S, Chen W. Evolview v3: a webserver for visualization, annotation, and management of phylogenetic trees 2019;47:270–5.
- Letunic I, Bork P. Interactive Tree Of Life (iTOL) v4: recent updates and 2019;47:256–9.
- Letunic I. SMART: recent updates, new developments and status in 2020 2021;49:2020–2.
- Marchler-bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, et al. CDD: NCBI’s conserved domain database 2015;43:222–6.
- Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, et al. The Pfam protein families database: towards a more sustainable future 2016;44:279–85.
- Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview Version 2 — a multiple sequence alignment editor and analysis workbench 2009;25:1189–91
- Bailey T and, Elkan C. Fitting a mixture model by expectation maximization. AAAI Press 1994:3–9.
- Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A(In) John M. Walker (ed): The Proteomics Protocols Handbook, Humana Press (2005).
- Hiller K, Grote A, Maneck M, Mu R, Jahn D. two-dimensional gel electrophoresis under consideration of membrane and secreted proteins 2006;22:2441–3.
- Yu C, Chen Y, Lu C, Hwang J. Prediction of Protein Subcellular Localization 2006;651:643–51.
- Horton P, Park K, Obayashi T, Fujita N, Harada H, Nakai K. WoLF PSORT: protein localization predictor 2007;35:585–7.
- Sperschneider J, Catanzariti A, Deboer K, Petre B, Gardiner DM, Singh KB, et al. LOCALIZER: subcellular localization prediction of both plant and effector proteins in the plant cell. Nat Publ Gr 2017:1–14.

{kind=link}