
Capítulo de livro publicado no livro do I CONGEB. Para acessa-lo clique aqui.
DOI: https://doi.org/10.53934/9786585062046-14
Este trabalho foi escrito por:
Milenna Machado Pirovani*; Renata Torezani ; Greiciane Gaburro Paneto
*Autor correspondente (Corresponding author) – Email: [email protected]
O novo coronavírus teve o seu início na cidade chinesa de Wuhan, no ano de 2019, e logo se tornou um vírus emergente responsável por levar a humanidade a uma grave crise sanitária global. O nome do vírus que causa a doença é SARS-CoV-2. A diversidade deste acontece, principalmente, por três motivos já estudados: (I) A enzima RNA polimerase, dependente de RNA, responsável pela replicação do seu material genético, não segue a sequência genética com total fidelidade; (II) como consequência do primeiro item, este possui frequência elevada de recombinação homóloga do RNA; (III) os coronavírus possuem genomas grandes, sendo que o seu tamanho pode variar de 26 a 31 kb. Então, por conta desses motivos, há uma grande capacidade para se mutar. Dessa forma, o objetivo deste trabalho foi, por meio da bioinformática, detectar mutações do SARS-CoV-2 presente em amostras de residentes do estado do Espírito Santo. As amostras foram obtidas a partir do banco de dados GISAID e alinhadas e analisadas com auxílio de softwares e linguagem de programação em Python. Até maio de 2022 foi possível obter 1990 amostras, destas, os sítios que mais sofreram mutações foram: 241, 3037, 14408, diversos sítios na região spike, 28278, 28881, 28882, 28883, além das inserções: AACA. Mutações nestes sítios comumente ocorrem ao redor do mundo. A maioria das amostras pertenciam a variante Delta, mas também há números significantes da variante Gamma, Zeta e Omicron. Tratou-se de um estudo in silico e os resultados deste estudo contribuem para o melhor entendimento da pandemia no Espírito Santo.
Palavras–chave: Bioinformática; Coronavírus; Mutação; Pandemia; SARS-CoV-2
INTRODUÇÃO
O novo coronavírus teve o seu início na China, no ano de 2019, na cidade de Wuhan. Se tornou responsável por uma emergência sanitária que levou a humanidade a uma grave crise sanitária global. Em poucos meses, o número de casos foram subindo rapidamente nos países asiáticos, que ultrapassou a barreira de outros continentes, evoluindo para uma Emergência de Saúde Pública de Importância Internacional, decretada pela Organização Mundial de Saúde (OMS) no dia 30 de janeiro de 2020 e, posteriormente, para uma pandemia no dia 11 de março de 2020 (1; 2). O primeiro caso de COVID-19 que foi confirmado no Brasil ocorreu no dia 26 de fevereiro de 2020 (3). Atualmente, em 2022, os casos confirmados no Brasil já ultrapassaram o número de 13 milhões (4).
Conforme houve a evolução de casos pelo mundo, estudos demonstraram que o vírus da Síndrome Aguda Respiratória Grave (SARS-CoV-2), causador da doença covid-19, sofria mutações muito frequentes (5; 6). A diversidade do coronavírus (CoV) acontece, principalmente, por três motivos já estudados: (I) A enzima RNA polimerase, dependente de RNA, responsável pela replicação do seu material genético, não segue a sequência genética com total fidelidade, o que gera elevadas taxas de mutação para os nucleotídeos replicados (o que os torna plásticos); (II) como consequência do primeiro item, o CoV possui frequência elevada de recombinação homóloga do RNA; (III) Os CoVs possuem genomas grandes, sendo que o seu tamanho pode variar de 26 a 31 kb. Isso contribui para uma maior plasticidade. Por conta desses três motivos, os CoVs foram levados a uma maior diversidade de genótipos e cepas, além de novas espécies que possuem a capacidade de se adaptar a novos nichos ecológicos e hospedeiro, conforme aconteceu com a pandemia causada pelo SARS-CoV-2, que foi um grande surto zoonótico (7; 8).
O genoma do SARS-CoV-2 codifica proteínas estruturais e não estruturais. As principais proteínas estruturais são: a proteína spike, também conhecida como proteína S, proteína de envelope, também chamada de proteína E, proteína de nucleocapsídeo (proteína N) e a proteína de membrana (proteína M). O envelope viral é formado pela proteína E e M, enquanto que a proteína N forma o conteúdo genético. O genoma tem duas regiões importantes que representam 2/3 de todo seu tamanho, o ORF1a e ORF1b. É nessas regiões que as proteínas não estruturais, também responsáveis pela replicação viral, são codificadas. São justamente essas proteínas acessórias que interrompem a resposta imune inata do hospedeiro (9).
Além dessas proteínas estruturais, o SARS-CoV-2 também possui proteínas não estruturais (NSPs), que são ativadas após a entrada do vírus na célula, a partir de poliproteínas de proteases virais. Essas proteases virais tem a função de liberar os NSPs para que o mesmo faça seu trabalho intracelular. Ao todo são 16 NSPs (9).
A bioinformática vem sendo utilizada para diversas análises nos últimos anos, incluindo a genômica, transcriptômica, proteômica, ciências da saúde e desenvolvimento de novos fármacos. O termo “bioinformática” é bastante amplo, abordando conhecimentos das ciências biológicas e da ciência da computação, utilizando linguagens de programação, como o R, Python e Perl, ou utilizando softwares ou banco de dados que foram desenvolvidos para esse tipo de análise (10; 11; 12; 13; 14). Desse modo, por facilitar a análise de variantes e mutações do SARS-CoV-2, o objetivo deste trabalho foi utilizar as ferramentas da bioinformática para detectar mutações do vírus causador da covid-19 presente em amostras de indivíduos residentes no estado do Espírito Santo.
MATERIAL E MÉTODOS
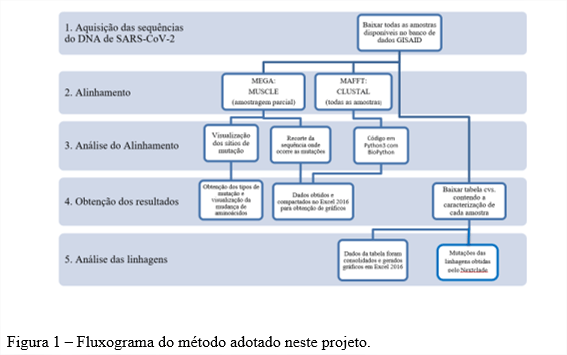
O trabalho ocorreu em 5 etapas, como consta na figura 1. A primeira etapa foi a aquisição das sequências, por meio do banco de dados GISAID (https://www.gisaid.org/). Foram selecionadas apenas amostras do estado do Espírito Santo, de genomas completos depositados no período de 01 de janeiro de 2020 a 20 de maio de 2022. A sequência de referência do vírus SARS-CoV-2 de Wuhan (China) foi obtida no banco de dados do National Center for Biothecnology Information (NCBI), depositada por Wu et al. (2020). A segunda etapa constou no alinhamento das sequências, que foi dividido em duas partes: inicialmente fez-se um alinhamento no software MEGA (Molecular Evolutionary Genetic Analysis, https://www.megasoftware.net/) pelo algoritmo MUSCLE, com uma amostragem parcial, objetivando a visualização dos SNPS e GAPS mais frequentes, que era anotado um recorte da sequência de códons nos quais as mutações ocorriam. Nesta etapa também foi possível visualizar se uma mutação era sinônima ou não (e posteriormente este dado foi confrontado com script em Python).
Para o alinhamento com todas as amostras foi utilizado o MAFFT version 7 online (https://mafft.cbrc.jp/alignment/server/ ), que é relativamente mais rápido que o MEGA. O alinhamento pelo MAFFT utilizou-se o Clustal e, igualmente ao MEGA, as amostras foram alinhadas juntamente com a amostra referência.
Após realizado os alinhamentos, ocorreu a análise dos mesmos. A análise no MEGA ocorreu de forma mais visual e manual, enquanto que o alinhamento gerado pelo MAFFT foi submetido em um script em python por meio da biblioteca BioPython version 1.79 (2021), utilizando o Jupyter Notebook (https://jupyter.org/) como ambiente de desenvolvimento integrado (IDE – Integrated Development Environment). O script pode ser visualizado na figura 2. O recorte das sequências dos códons nos quais ocorriam as mutações também foram implementados neste script para a verificação da frequência de mutação em todas as amostras. As saídas deste script foram copiadas para uma planilha Excel 2016 e os dados foram consolidados em gráficos.
Por fim, para análise das linhagens, que ocorreu após a análise do alinhamento, foi adquirida uma tabela que a própria plataforma GISAID oferece, contendo as linhagens de cada sequência depositada no banco de dados, que contém uma coluna para a nomenclatura oficial da OMS (Gamma, Delta, Omicron, etc.) e outra coluna com as nomenclaturas disponíveis no Pango (https://cov-lineages.org/). Os dados desta tabela foram consolidados no Excel 2016, analisados e gerados gráficos.

RESULTADOS E DISCUSSÃO
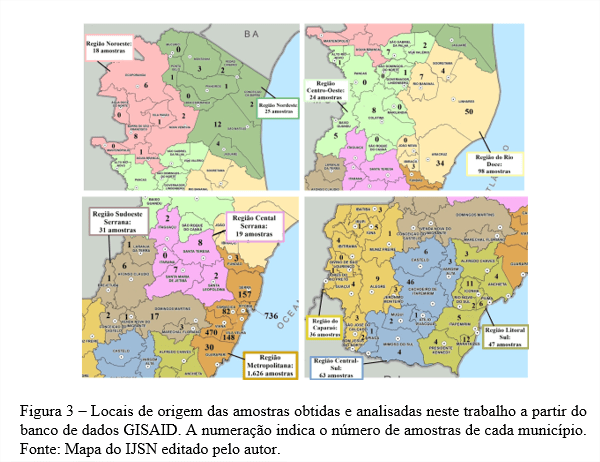
Foram obtidas e analisadas 1990 (hum mil e novecentos e noventa) amostras do genoma completo de SARS-CoV-2 de pessoas infectadas residentes do estado do Espírito Santo, disponíveis no banco de dados GISAID, de 01 de janeiro de 2020 até o dia 20 de maio de 2022. Além destas, também foi obtida a amostra referência de Wuhan, China, para alinhamento, totalizando 1991 (mil e novecentos e noventa e uma) amostras. Destas, 18 pertenciam a região Noroeste do estado, 25 da Nordeste, 24 da Centro-oeste, 98 da região do Rio-Doce, 31 da Sudoeste-serrana, 19 da Central-serrana, 1626 da Metropolitana, 36 da região do Caparaó, 63 da Central-Sul e 47 da região do Litoral-Sul. Das 1990 amostras, apenas 3 não continham informações do local de coleta, apenas relatando serem do estado do Espírito Santo (Figura 3).
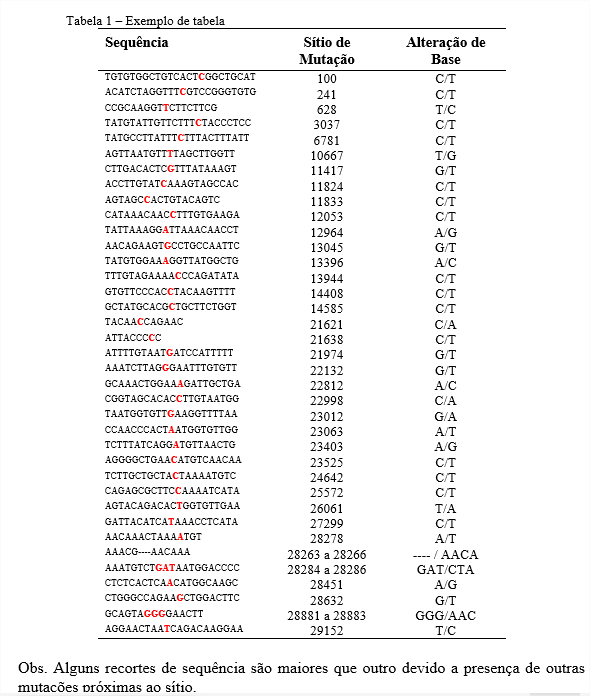
Após as amostras serem obtidas do banco de dados, as mesmas foram submetidas ao alinhamento pelo software MEGA. Por este alinhamento foi possível observar algumas mutações em sítios específicos. Na figura 4, cada linha pontilhada equivale a uma amostra obtida no GISAID, enquanto que a sequência visualizada no topo da imagem equivale à amostra original (referência) obtida no NCBI. Os pontos significam que a sequência se mantém igual à que está no topo. Pode-se visualizar que a maioria das amostras apresentaram mutação de C/T no sítio 3037 (nesta imagem). Ao todo, foram identificadas 37 mutações por este software, que estão destacadas em vermelho na tabela 1.

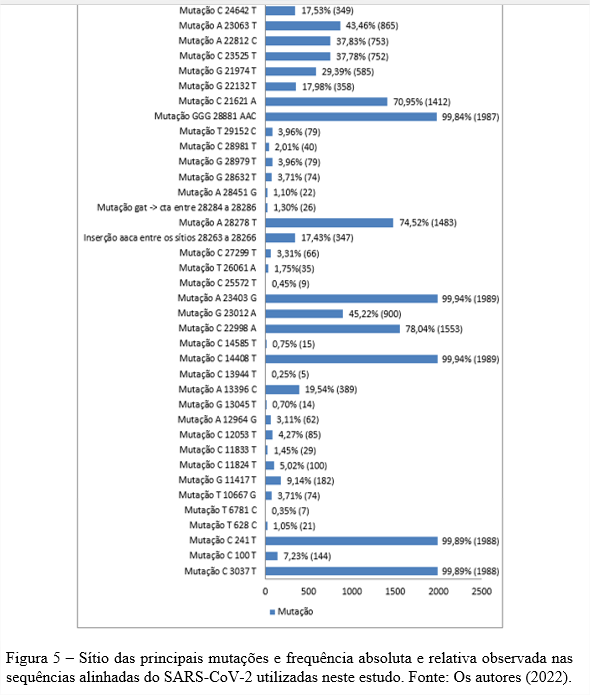
Os sítios que mais obtiveram mutações foram: 241, 3037, 14408, 21621, 21974, 22132, 22812, 22998, 23012, 23063, 23403, 23525, 24642, 28278, 28881, 28882, 28883 e a inserção AACA também foi frequente em quase 20% das amostras (347 amostras), entre os sítios 28263 a 28266. A contagem das mutações pode ser visualizada na figura 5. Destas mutações, 70,27% eram não-sinônimas, 24,32% sinônimas e 5,40% houve a mudança da trinca de nucleotídeos para um stop códon. Tanto a mutação C241T quanto a C3037T são exemplos de mutações sinônimas (15). C241T trata-se de uma mutação de alta frequência, que já foi identificada no mundo inteiro, apresentando frequência global superior a 95%, o que é compatível com os resultados apresentados neste trabalho, já que 99,89% das amostras apresentavam mutação neste sítio. Apesar de ser uma mutação sinônima, são descritos efeitos secundários na estrutura do RNA, o que influencia na taxa de replicação e, por conseguinte, na infecção viral. Não há mudança significativa na estrutura secundária, porém a mutação C241T altera uma alça de haste (SL4), que em situações de SNP simples ou duplo, pode reduzir a replicação viral (16; 17). Além disso, esta mutação também está associada aos clados 20A e 20B (18). A mutação C100T também se encontra nesta região 5’UTR e está bastante associada à linhagem B.1.1.28 (19; 20).
Já em relação à mutação C3037T, apesar de sua frequência global ser maior que a C241T, já que esta mutação causa uma mudança de códon (TTC>TTT) sendo mais frequente na cepa do SARS-CoV-2 (21; 17), ainda não há muitos estudos que mostram exatamente qual o seu impacto. Existem algumas hipóteses de que esta mutação, em associação com a C241T, C14408T e A23403G, influenciam em sua patogenicidade, infectividade ou na adaptabilidade ao hospedeiro do SARS-CoV-2, além disso, todas essas mutações são características do clado G. Ademais, o sítio 3037 foi exposto como sítio hipermutável de baixa adequação, juntamente com o 11074 (22; 17; 23).
A mutação C14408T resulta em uma alteração em P323 para leucina de RNA polimerase dependente de RNA. Esta por sua vez, tem função, juntamente com a P322, de terminar a hélice 10 e gerar uma volta seguida por uma folha beta. A leucina nesta posição forma interações hidrofóbicas que cria uma variante de maior estabilidade para NSP12. Esta mutação está associada à evolução do genoma e maior densidade de mutação, podendo estar agindo de forma sinérgica com a mutação A 23403 G (24; 21). Já a mutação A 23403 G resulta em uma mutação da proteína spike D 614 G. Esta mutação está associada ao aumento da infecção viral, pela lógica de que quanto menos houver liberação de spikes, maior será a incorporação em pseudovírus e aumento da carga viral. Diversos experimentos in vitro conseguiram provar a infectividade desta mutação, o que pode ser levado em consideração para o desenvolvimento de vacinas a base de RNA (25; 26; 23).
A mutação G28881A e G28882A são não-sinônimas, mudando uma arginina para uma lisina e a mutação G28883C troca uma glicina por uma arginina, isso favorece para uma carga mais positiva à proteína N, havendo a hipótese de que essas mutações podem modificar a flexibilidade molecular desta proteína (21; 27). Inclusive, relatos de que a mutação G28883C (G 204 R) gera uma mutação de nucleocapsídeo capaz de aumentar a infectividade e virulência do SARS-CoV-2 (28). A proteína N tem função estrutural e essencial para o vírus, pois realiza a regulação do metabolismo celular infectado e no empacotamento do genoma viral, possuindo função importante na replicação e transcrição (29).
As mutações não-sinônimas nos sítios: C21621A (T20N), C21638T (P26S), G21974T (D138Y), G22132T (R190S), C23525T (H655Y), A22812C (K417T), G23012A (E484K), A23063T (N501Y), C24642T (T1027I) são todas na região da proteína S.
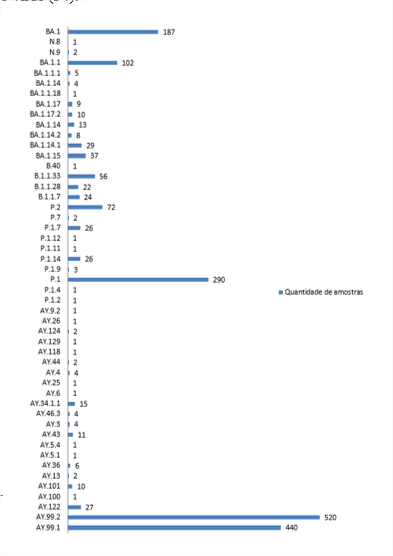
Das amostras analisadas, a maioria das que foram obtidas pelo GISAID pertenciam à linhagem AY.99.2 e AY.99.1 (ambas delta), sendo 26,13% e 22,11%, respectivamente. Existem também muitas amostras pertencentes às linhagens P.1 (Gamma (14,57%)), BA.1 (Omicron (9,39%)) e BA.1.1 (Omicron (5,12%)), sendo estas a de maiores números, porém também foram encontradas amostras com as linhagens: AY.100, AY.101, AY.122, AY.13, AY.36, AY.5.1, AY.5.4, AY.43, AY.3, AY.46.3, AY.34.1.1, AY.6, AY.25, AY.4, AY.44, AY.118, AY.129, AY.124, AY.26, AY.9.2, P.1.2, P.1.4, P.1.9, P.1.14, P.1.11, P.1.12, P.1.7, P.2, P.7, B.1.1.7, B.1.1.28, B.1.1.33, B.40, BA.1.15, BA.1.14.1, BA.1.14.2, BA.1.14, BA.1.17, BA.1.17.2, BA.1.1.18, N.9 e N.8 (Figura 6).
A subdivisão de linhagem AY.99.2 possui mutação em A1306S, P2046L, V2930L, T3255I, T3646A, T4087I em ORF1a, P314L, G662L, A1918V em ORF1b, T19R, L452R, T478K, D614G, D950N em S, S26L em ORF3, I82T em M, V82A, T120I em ORF7a, G215C em N (30). Um estudo realizado no Rio de Janeiro também relatou dominância das sublinhagens AY.99.2 em relação a AY.99.1 (o que também é visualizado neste trabalho) (31). O AY.99.2 difere do AY.99.1 pela mutação C4927T (32).
A linhagem P.1 possui algumas mutações na proteína S que são características, como L18F, T20N, P26S, D138Y, R190S, K417T, E484K, N501Y, H655Y e T1027I quando comparada com a sua ancestral B.1.1.28 (33). A BA.1 possui 11 mutações que são compartilhadas também em suas sublinhagens, como a G339D, S373P, S375F, K417N, N440K, S477N, T478K, E484A, Q4934, Q498R e N501Y. Estas mutações contribuem de forma significativa na transmissão em potencial e evasão imune do hospedeiro do vírus (34).
CONCLUSÕES
Tratou-se de um estudo in silico, na qual as mutações encontradas foram coerentes com o que está descrito na literatura, principalmente os sítios de alta frequência global (C100T, C241T, C3037T). Foram observados que muitas mutações ocorreram na região da proteína S, assim como tem sido estudado desde o início da pandemia. Existem algumas mutações que são descritas como características de certas linhagens, como a mutação não-sinônima T20N (C21621A), associada a linhagem P.1 (Gamma), porém, neste estudo, esta mutação foi encontrada em mais amostras que correspondiam a esta linhagem (70,95% das amostras continham esta mutação, mas apenas 14,57% das amostras pertenciam a linhagem P.1). O mesmo ocorre com a N501Y (A23063T), descrita como característica da linhagem P.1, porém com uma frequência bem maior quando analisada a quantidade de amostras que são provenientes desta linhagem. Isto demonstra que estudos mais específicos a respeito da evolução do vírus e quais realmente caracterizam suas linhagens devam ser desenvolvidos.
Ademais, existem muitas mutações encontradas que ainda não há muito de suas origens e funcionalidades descritas em literatura. Apesar de que a maioria não apresenta uma frequência alta em relação ao número de amostras, isto deve ser reconsiderado, já que as mutações podem interferir diretamente à eficácia de vacinas, terapias utilizadas para a covid-19 e até mesmo ao desenvolvimento da doença no paciente.
AGRADECIMENTOS
A todos os colaboradores e cientistas que depositaram os genomas no banco de dados GISAID, que foi vital para o desenvolvimento deste estudo.
REFERÊNCIAS
- World Health Organization: WHO. Statement on the meeting of the International Health Regulations (2005) Emergency Committee regarding the outbreak of novel coronavirus (2019-nCoV) [Internet]. Who.int. World Health Organization: WHO; 2020. Available from: https://www.who.int/news-room/detail/23-01-2020-statement-on-the-meeting-of-the-international-health-regulations-(2005)-emergency-committee-regarding-the-outbreak-of-novel-coronavirus-(2019-ncov)
- WHO. WHO Director-General’s opening remarks at the media briefing on COVID-19 – 11 March 2020 [Internet]. www.who.int. 2020. Available from: https://www.who.int/dg/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19—11-march-2020
- Candido DDS, Watts A, Abade L, Kraemer MUG, Pybus OG, Croda J, et al. Routes for COVID-19 importation in Brazil. Journal of Travel Medicine. 2020 Mar 23;27(3).
- Ministério da Saúde. Coronavírus Brasil [Internet]. covid.saude.gov.br. Available from: https://covid.saude.gov.br/
- Benito LAO, Lima R da C, Palmeira AM de L, Karnikowski MG de O, Silva ICR da. Variantes do vírus SARS-COV-2 causadoras da COVID-19 no Brasil. Revista de Divulgação Científica Sena Aires. 2021 Mar 28;205–19.
- Freitas ARR, Giovanetti M, Alcantara LCJ. Emerging variants of SARS-CoV-2 and its public health implications. InterAmerican Journal of Medicine and Health [Internet]. 2021 Feb 8 [cited 2021 May 17];4. Available from: https://www.iajmh.com/iajmh/article/view/181
- Woo PC, Lau SK, Yuen K. Infectious diseases emerging from Chinese wet-markets: zoonotic origins of severe respiratory viral infections. Current Opinion in Infectious Diseases. 2006 Oct;19(5):401–7.
- Woo PCY, Lau SKP, Huang Y, Yuen K-Y. Coronavirus Diversity, Phylogeny and Interspecies Jumping. Experimental Biology and Medicine. 2009 Oct;234(10):1117–27.
- Mallick R, Duttaroy AK. Structural, molecular biology and the immunopathology of SARS-CoV-2: An updated review. 2020 Jul 6.
- Ferrari RC, Gastaldi, VD. Precisamos falar sobre Bioinformática. Universidade de São Paulo: VIII Botânica no Inverno. Capítulo 20, p. 246-262. 2018.
- Lehugeur T de P, Melo HCS. BIOINFORMÁTICA APLICADA NO DESENVOLVIMENTO DE NOVOS FÁRMACOS. Psicologia e Saúde em debate [Internet]. 2018 Dec 12 [cited 2022 Aug 23];4(Suppl1):55–5. Available from: http://www.psicodebate.dpgpsifpm.com.br/index.php/periodico/article/view/401
- Andrade JV, Colucci YS, Gonçalves MG, Simões JPC, Carneiro DO, Shibuya CM, Siqueira AP. Participação em um curso de Bioinformática Aplicada a Oncologia Molecular: Relato de Experiência. Sistema eletrônico de administração de conferências. Vol. IV. 2019. Avaible from: https://conferencia.fagoc.br/index.php/trabalhosfagoc/MOSTRAUNIFAGOCIV/paper/view/1072
- Lourenço DA, Choupina A. Bioinformática aplicada à caracterização de íntrons. Adolescência: Revista Júnior de Investigação [Internet]. 2019 [cited 2022 Aug 23];6(1):62–70. Available from: https://bibliotecadigital.ipb.pt/handle/10198/20452
- Pereira NG, Oliveira LF, Ortega JR, Versiani MS, Oliveira AME, Xavier AREO, Xavier, MAS. Bioinformática Como Ferramenta Na Análise De Epitopos Antigênicos No Design De Vacinas Contra Anaplasma Marginale, Leishmania Spp., Sars-Cov-2 E Toxina De Clostridium Septicum. BJD. 2019; 7(4).
- Yang X, Dong N, Chan EW-C, Shen S. Identification of super-transmitters of SARS-CoV-2. medRxiv. 2020. 144-149.
- Picot S, Marty A, Bienvenu A-L, Blumberg LH, Dupouy-Camet J, Carnevale P, et al. Coalition: Advocacy for prospective clinical trials to test the post-exposure potential of hydroxychloroquine against COVID-19. One Health. 2020 Apr;100131.
- Mercatelli D, Giorgi FM. Geographic and Genomic Distribution of SARS-CoV-2 Mutations. Frontiers in Microbiology. 2020 Jul 22;11.
- Raghav S, Ghosh A, Turuk J, Kumar S, Jha A, Madhulika S, et al. Analysis of Indian SARS-CoV-2 Genomes Reveals Prevalence of D614G Mutation in Spike Protein Predicting an Increase in Interaction With TMPRSS2 and Virus Infectivity. Frontiers in Microbiology. 2020 Nov 23;11.
- de Souza UJB, dos Santos RN, Campos FS, Lourenço KL, da Fonseca FG, Spilki FR. High Rate of Mutational Events in SARS-CoV-2 Genomes across Brazilian Geographical Regions, February 2020 to June 2021. Viruses. 2021 Sep 10;13(9):1806.
- Phylogenetic relationship of SARS-CoV-2 sequences from Amazonas with emerging Brazilian variants harboring mutations E484K and N501Y in the Spike protein [Internet]. Virological. 2021. Available from: https://virological.org/t/phylogenetic-relationship-of-sars-cov-2-sequences-from-amazonas-with-emerging-brazilian-variants-harboring-mutations-e484k-and-n501y-in-the-spike-protein/585
- Justo Arevalo S, Zapata Sifuentes D, Huallpa CJ, Landa Bianchi G, Castillo Chávez A, Garavito-Salini Casas R, et al. Global Geographic and Temporal Analysis of SARS-CoV-2 Haplotypes Normalized by COVID-19 Cases During the Pandemic. Frontiers in Microbiology. 2021 Feb 17;12.
- Demir Ab, Benvenuto D, Abacioğlu H, Angeletti S, Ciccozzi M. Identification of the nucleotide substitutions in 62 SARS-CoV-2 sequences from Turkey. TURKISH JOURNAL OF BIOLOGY. 2020 Jun 21;44(3):178–84.
- Xi B, Jiang D, Li S, Lon JR, Bai Y, Lin S, et al. AutoVEM: An automated tool to real-time monitor epidemic trends and key mutations in SARS-CoV-2 evolution. Computational and Structural Biotechnology Journal. 2021;19:1976–85.
- Eskier D, Karakülah G, Suner A, Oktay Y. RdRp mutations are associated with SARS-CoV-2 genome evolution. PeerJ [Internet]. 2020 Jul 21 [cited 2020 Oct 1];8. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7380272/
- Biswas SK, Mudi SR. Spike protein D614G and RdRp P323L: the SARS-CoV-2 mutations associated with severity of COVID-19. Genomics & Informatics. 2020 Dec 31;18(4):e44.
- Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, et al. Tracking changes in SARS-CoV-2 Spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell [Internet]. 2020 Jul;182(4). Available from: https://www.cell.com/cell/fulltext/S0092-8674(20)30820-5
- Ugurel OM, Ata O, Turgut-Balik D. An updated analysis of variations in SARS-CoV-2 genome. TURKISH JOURNAL OF BIOLOGY. 2020.
- Wu H, Xing N, Meng K, Fu B, Xue W, Dong P, et al. Nucleocapsid mutations R203K/G204R increase the infectivity, fitness, and virulence of SARS-CoV-2. Cell Host & Microbe [Internet]. 2021 Dec 8 [cited 2022 Mar 24];29(12):1788-1801.e6. Available from: https://pubmed.ncbi.nlm.nih.gov/34822776/#:~:text=Our%20work%20suggests%20that%20the
- Kang S, Yang M, Hong Z, Zhang L, Huang Z, Chen X, et al. Crystal structure of SARS-CoV-2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. Acta Pharmaceutica Sinica B. 2020 Apr;10(7).
- Gularte JS, da Silva MS, Mosena ACS, Demoliner M, Hansen AW, Filippi M, et al. Early introduction, dispersal and evolution of Delta SARS-CoV-2 in Southern Brazil, late predominance of AY.99.2 and AY.101 related lineages. Virus Res [Internet]. 2022 [cited 2022 Aug 23];198702–2. Available from: https://pesquisa.bvsalud.org/global-literature-on-novel-coronavirus-2019-ncov/resource/pt/covidwho-1655224
- Lamarca AP, Almeida LGP de, Francisco Junior R da S, Cavalcante L, Brustolini O, Gerber AL, et al. Phylodynamic analysis of SARS-CoV-2 spread in Rio de Janeiro, Brazil, highlights how metropolitan areas act as dispersal hubs for new variants. 2022 Jan 17.
- Romano CM, de Oliveira CM, da Silva LS, Levi JE. Early Emergence and Dispersal of Delta SARS-CoV-2 Lineage AY.99.2 in Brazil. Frontiers in Medicine [Internet]. 2022 [cited 2022 Jul 7];9:930380. Available from: https://pubmed.ncbi.nlm.nih.gov/35783651/
- Faria NR, Mellan TA, Whittaker C, Claro IM, Candido D da S, Mishra S, et al. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science. 2021 Apr 14;eabh2644.
- Kumar S, Karuppanan K, Subramaniam G. Omicron (BA.1) and sub‐variants (BA.1.1, BA.2, and BA.3) of SARS‐CoV‐2 spike infectivity and pathogenicity: A comparative sequence and structural‐based computational assessment. Journal of Medical Virology. 2022 Jun 16;

{kind=link}